Hypohalous Acids
The most common hypohalous acids, HOCl and HOBr, are relatively weak acids (pKa = 7.5 & 8.6 respectively) that are only stable in solution. These acids and their ester derivatives are formed reversibly when the corresponding halogens are dissolved in hydroxylic solvents.
| X2 + H2O |  |
HOX + HX |
|---|---|---|
| X2 + ROH | |
ROX + HX |
For a solution of chlorine in water the equilibrium constant is only about 5 * 10-4. The concentration of HOCl may be increased by adding silver oxide ( Ag(+) reduces the chloride ion concentration and the oxide increases the pH ), but this is not a common procedure. As a rule, these reagents are prepared by hydrolysis of N-chloro and N-bromo precursors immediately prior to their use.
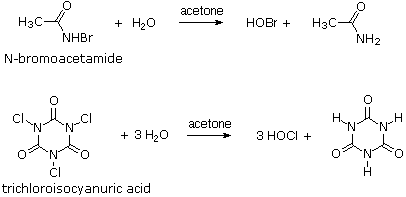 |
|---|
End of this supplementary topic
Writing Mechanisms
Writing Reaction Mechanisms
When a chemical reaction takes place, the atoms and associated electrons
of the reacting molecular species reorganize to form new product molecules
and ions. The overall structural changes are normally evident once the
products have been identified, but the manner in which these changes take
place is less obvious. In order to represent and keep track of the electron
reorganization involved in a reaction, chemists find it useful to depict
the way in which key electron pairs (or single electrons) move to
accommodate the breaking of existing bonds and the making of new bonds.
Curved
arrow symbols are a convenient tool for this purpose, and a complete
representation of all the electron shifts in a reaction is called its
mechanism.
As a rule, only the valence shell electrons of atoms are involved in a
chemical reaction, and only a few of these electrons actually change their
location. Electron pair shifts are the most common events in most
mechanisms, and are the subject of this discussion. Single (or odd)
electron shift mechanisms are described in the Free Radical section
of this text. Valence shell electron pairs will either be bonding or
non-bonding. The placement of curved arrows for these pairs will be
slightly different. This is illustrated by the two equations in the
following diagram. Non bonding electron pairs usually participate in a
reaction by forming a covalent bond to another atom. Bonding electron pairs
may shift to form a new covalent bond, or to become a non-bonding electron
pair. By clicking on this diagram, these changes will be identified by
color coding.
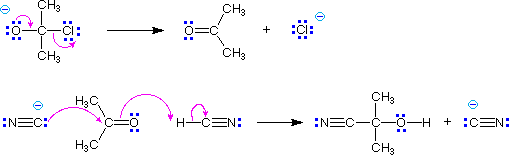
In these examples the foot of the curved arrow (the end opposite to the head) is placed at the initial location of an electron pair that is going to shift its position. If this is a bonding electron pair that should be the center of the bond. The head of the arrow then points to the pair's final location – either an atom if it becomes non-bonding, or the space where a new bond will be formed. If the new bond will be a π-bond, this should be the center of the already existing σ-bond. If the new bond will be a σ-bond, the arrow should clearly identify the atom that will share the electron pair.
In cases where full or partial charges are present, the direction in which arrows are drawn should be evident. Remember, a curved arrow shows the movement of an electron pair in the direction to which the head points, and nucleophiles generally bond to electrophiles. The previous two equations are typical examples. In other cases, especially when a cycle of arrows is drawn, the direction may not be clear and may not even matter. Two examples of such cyclic arrays are shown in the following equations. The first describes the resonance π-electron delocalization in benzene. The second is a Diels-Alder cycloaddition reaction between symmetrically substituted reactants.
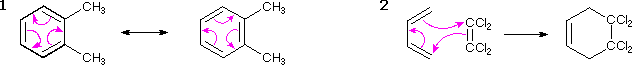
In reactions where the distribution of electrons is not symmetrical, the direction in which curved arrows are drawn may be significant. One such case is the thermal decarboxylation of acetoacetic acid shown below. A concerted mechanism consisting of cyclic shifts of electron pairs may be drawn, and two such examples are shown below. Although both mechanisms lead to the products in the light blue box, the mechanism on the left (green arrow) is a more plausible representation than that on the right, because it reflects the acidic character of the carboxylic acid and the basic character of a carbonyl oxygen.
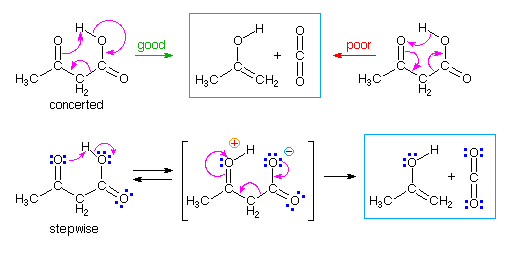
This distinction is clearly seen in the two-step mechanism written in the second row of the diagram. The initial proton transfer proceeds in the expected fashion, and the remaining electron shifts are the same as those in the green arrow concerted mechanism. There is no evidence supporting a two-step pathway for this decarboxylation, but it is possible that the proton transfer stage leads the concerted cyclic sequence.
The preceding example is nevertheless a warning that many reactions do take place in a stepwise fashion, and the mechanisms we write must recognize such cases. The conversion of an ester to an amide by reaction with ammonia, as shown below, is such an example. At first glance this seems to be a substitution reaction, and it would be possible to draw curved arrows for a concerted mechanism, as depicted in the box on the far right. In fact, this reaction, and other acyl transfer reactions, take place by an addition-elimination sequence, as drawn in the boxed space beneath the overall reaction equation. The concerted mechanism on the right is therefore an erroneous conjecture. Thus we have a useful warning that "arrow pushing" does not always lead to plausible interpretations of reaction mechanisms. Experimental support for any mechanism is highly desirable if not essential.
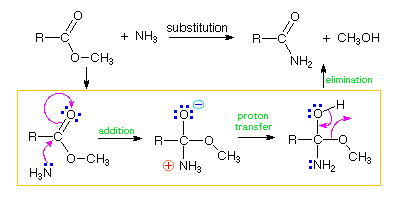 |
 This is not a plausible mechanism |
|---|
|
Oxford University. |
End of this supplementary topic
Structure & Acidity
Molecular Structure and Acidity
1. Equilibrium Acidity
pKa Acidities of Some Common Hydrides
| 4 | 5 | 6 | 7 |
|---|---|---|---|
| CH3-H ca. 50 |
NH2-H 34 |
HO-H 15.74 |
F-H 3.2 |
| HS-H 6.97 (pK1) |
Cl-H -3 |
||
| HSe-H 3.8 (pK1) |
Br-H -6 |
||
| HTe-H 2.6 (pK1) |
I-H -7 |
The relative acidities of different acids are commonly measured and cited as pKa values, relative to a standard solvent base, often water. These numbers reflect the equilibrium acidities of the acids. An astounding range of acidities is displayed by even rather simple compounds. The table on the right lists some elemental hydrides from groups 4 through 7 of the periodic table. The pKa's determined (or in some cases estimated) for these compounds are shown beneath the formulas. Approximate values for higher members of group 4 and 5 hydrides (e.g. silane and phosphine) have not been reported. Note that these logarithmic numbers encompass nearly sixty powers of ten. This is a greater span than that encompassed by distance measurements starting from the radius of a hydrogen atom and extending to the diameter of the known universe.
Why do these relatively simple compounds differ in acid strength so
markedly? Two factors may be discerned:
• First, the compounds in the top row clearly show the importance of
electronegativity.
All the heavier elements have greater electronegativities than hydrogen,
with carbon being the least different. The ionic character of these
covalent bonds is such that hydrogen carries a partial positive charge, and
the heavier atom a corresponding negative charge. The greatest charge
separation is in H-F, where the electronegativity difference is nearly 2.
Removal of a proton is facilitated by this charge separation. The covalent
bond energies do not correlate inversely with acid strength, as one might
have expected, since the two strongest acids have the strongest bonds (H–O
111 kcal/mol & H–F 135 kcal/mol). Finally, the heavy atoms in the top
row have similar sizes, the covalent radii being 0.75 ±0.02 Å. The
importance of this fact will become apparent in the next discussion.
• Second, the compounds in the columns representing periodic groups 6 and 7
show an increase in acidity moving from the top to the bottom. This is
opposite to the electronegativity change, and is best attributed to an
increase in heavy atom size. When an acid transfers a proton to a base, the
remaining residue (the conjugate base) must carry a negative charge.
Ignoring solvent stabilization (solvation),
the stability of ions is a function of charge density. A small ion has a
higher charge density than a larger ion of the same charge, making the
smaller ion less stable. From the covalent radius of oxygen compared with
sulfur, and fluorine compared with chlorine, it can be estimated that the
charge density on the larger atom is half that of the smaller. The
resulting stabilization of the conjugate base more than compensates for the
decrease in electronegativity in moving down the column; so H2S
is a stronger acid than H2O, and HCl a stronger acid than HF.
Since sulfur and chlorine are nearly the same size ( covalent radii being
1.02 ±0.02 Å ), electronegativity explains the difference in acidity
between H2S and HCl.
| NH4(+) 9.24 |
OH3(+) -1.74 |
S-H(–) 15 (pK2) |
Se-H(–) 11 (pK2) |
If the heavy atom of an acid carries a formal charge, its acidity will be changed substantially. This is demonstrated by the examples on the right. Ammonium and hydronium ions carry a positive charge, and the acidity of the species is increased by over fifteen powers of ten relative to uncharged ammonia and water. By contrast, hydrogen sulfide and hydrogen selenide are dibasic acids (they have two acidic protons). Once the first proton has been lost, the acidity of the negatively charged conjugate base is reduced over a million fold. This is true for most other dibasic acids such as H2SO4 and H2CO3.
Accurate acidity measurements in the pKa range from 1 to 14 can usually be made in water solution. However, acids stronger than the hydronium ion (H3O(+)) and bases stronger than hydroxide ion (OH(–)) react immediately with this solvent, and the resulting "leveling effect" prevents direct measurement of their pKa's. One way of circumventing this difficulty is to examine the acidity of very strong ( pKa < 0) and very weak ( pKa > 15) acids in different (non-aqueous) solvents, and to extrapolate these measurements to water. For example, solvents such as acetic acid, acetonitrile and nitromethane are often used for studying very strong acids. Very weakly acidic solvents such as DMSO, acetonitrile, toluene, amines and ammonia are used to study the acidities of very weak acids. The errors introduced in extreme cases, such as methane, are often large; but the overall range of acid strengths observed in this manner cannot be questioned.
2. Solvent Effects
Acidity
As noted earlier, a Brønsted acid base equilibrium involves a reversible proton transfer between a pair of acids and a pair of bases (referred to as conjugate pairs). The dissociation of a group of acids in a given solvent may be used as a measure of acid strength, and in such cases the solvent serves as a common reference base. The determination of Ka and pKa values for any solvent system may be carried out in the same way as in water; however, the acidities obtained for a group of acids measured in different solvents will generally be different, both in the numerical value for each, and sometimes in relative order.
| A–H an acid |
+ | Sol: solvent |
 |
A:(–) conjugate base |
+ | Sol–H(+) conjugate acid |
There are several factors which influence acidity measurements made in water and other solvents. These are:
1. Ionization of an acid produces
ions. Solvents having large dielectric constants favor charge formation
and separation.
2. The conjugate acid from the solvent
is a cation that is stabilized by association with other solvent
molecules, a phenomenon called solvation.
3. The conjugate base of the acid is an
anion, that in most cases is also stabilized by solvation.
As examples, consider the following data obtained for six strong acids in three common solvents.
Experimental pKa Values for Some Strong Acids in Different Solvents
|
||||||||||||||||||||||||||||
| * Perchloric acid and trifluoromethanesulfonic acid are both
completely ionized in water and DMSO. The pKa value of –10 is approximate, and is intended to reflect the high degree of dissociation. |
All three solvents have fairly high dielectric constants, but water (ε = 80) is significantly greater than DMSO (ε = 46.7) or acetonitrile (ε = 37.5). The greater acidity ( lower pKa ) of the last four compounds in water might be attributed to the dielectric difference; however, the superior anion solvation provided by water is considered to be the major factor. Both DMSO and acetonitrile are poor anion solvation solvents, consequently the ionization equilibrium shown above will be shifted to the left.
It is informative to focus first on the pKa differences
between water and DMSO. We do this because water and DMSO have similar
basicities, so stabilization of the solvent conjugate acid species should
not differ substantially. HBr and HCl display pKas in DMSO that
are roughly 9 to 10 powers of ten more positive than in water, and the
weaker second period acid HF shows an even greater change (pKa =
3.17 in water & 15 in DMSO). The conjugate bases of these acids are all
single atom anions. Because the negative charge is localized, anion
stabilization by solvation is probably the most important factor in
establishing the position of these equilibria. In contrast, the conjugate
bases from sulfuric acid and trifluoroacetic acid are internally stabilized
by charge delocalization over two or three oxygen atoms. The importance of
solvation is therefore less important, and the shift in acidity in DMSO is
reduced to 4 powers of ten.
When acetonitrile is the solvent, the measured pKas of these
acids show a further increase, and even the strongest acids are not
completely dissociated. This change may be attributed in large part to
diminished solvation of the solvent conjugate acid, which is the chief
cation species in dilute solutions. Since both the cationic and anionic
species from acidic ionization are poorly stabilized by solvation, the
dissociation equilibrium lies far to the left (no ionization).
Most organic compounds are much weaker acids than those listed above. It
is therefore of interest to determine the changes in acidity that take
place when such compounds are examined in the same three solvents. The
first four compounds in the following table (shaded light green) are weak
acids having conjugate bases in which the charge is localized on a single
atom ( O, C & S ). Changing the solvent from water to DMSO decreases
the acidity by 10 to 12 powers of ten for second period atoms ( C & O
), and by 6.5 for the third period sulfhydryl acid. Just as in the more
acidic cases treated above, solvation of the conjugate base anions is an
important stabilizing factor in water, although the lower charge density on
sulfur reduces its significance. The further increase in pKa
that occurs in acetonitrile solutions, relative to DMSO, is fairly uniform
(10 to12 units) and a bit larger than the shift found for the strong
acids.
The last four compounds (shaded blue) have conjugate bases stabilized by
charge delocalization. Here, the decrease in acidity going from water to
DMSO is lowered to 7 to 8 powers of ten for oxygen and is less than 4 for
sulfur. As noted previously, charge delocalization within an anion reduces
the importance of solvation stabilization. The increase in pKa
found for acetonitrile solutions remains roughly the same, and may be
attributed to poor solvation of the nitrile conjugate acid, as noted
above.
Experimental pKas for Some Weak Acids in Different Solvents
| Compound | H2O | DMSO | CH3CN |
|---|---|---|---|
| C2H5OH | 17 | 29 | 42 |
| CF3CH2OH | 12.4 | 23.5 | 33.5 |
| C6H5C≡CH | 19 | 29 | 40.6 |
| C4H9SH | 10.5 | 17 | 28.6 |
| CH3CO2H | 4.8 | 12.8 | 22.3 |
| C6H5CO2H | 4.2 | 11.1 | 20.7 |
| C6H5OH | 10 | 18 | 26.6 |
| C6H5SH | 6.6 | 10.3 | 20.6 |
With the exception of the acetylene derivative, the previous compounds are all heteroatom acids. Similar measurements for a group of activated carbon acids show agreement with the previous analysis, and illustrate the manner in which anionic charge delocalization reduces the pKa difference between water and DMSO measurements. Note the near identity of the second and third examples in the following table. As expected, the pKa increase in going to acetonitrile from DMSO remains roughly constant (ca. 11).
Experimental pKas for Some Carbon Acids in Different Solvents
| Compound | H2O | DMSO | CH3CN |
|---|---|---|---|
| (CH3CO)2CH2 | 9 | 13.3 | 24.4 |
| (N≡C)2CH2 | 11.2 | 11.2 | 22.2 |
| (C6H5SO2)2CH2 | 11.6 | 12.2 | 23.2 |
| (C2H5OCO)2CH2 | 13.5 | 16.4 | 27.8 |
| C4H4CH2 cyclopentadiene |
15.0 | 18.0 | 31.2 |
Basicity
The most common notation for reporting relative base strengths is in terms of the pKa's of the corresponding conjugate acids ( these conjugate acids are often called "onium" cations ). The following equation shows the equilibrium involved in this relationship. Note that strong bases will have weakly acidic conjugate acids, so the pKa is proportional to the base strength of the base.
| B–H(+) conjugate acid |
+ | Sol: solvent |
|
B: a base |
+ | Sol–H(+) conjugate acid |
The previously defined factors that influenced acidity may now be reexamined for this new equilibrium.
1. Ions are present on both sides of
the equation. Consequently, differences in the solvent dielectric
constant should be a relatively unimportant factor.
2. The base and solvent conjugate acid
cations are both stabilized by solvation. This will be a critical factor,
and is related to solvent basicity.
3. There are no anions in the above
equation; however charge neutralization requires a counter anion. Since
the same anion will be present on both sides of the equation, its
influence on this equilibrium will be canceled.
The following table gives pKa values for some commonly used
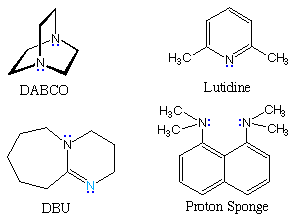
nitrogen bases, some structures for which are shown to its right. In the
triethylamine and DABCO examples at the top of the table, the cationic
charge is relatively localized on a single nitrogen atom. The charge in
lutidine may be delocalized onto ring carbons at the cost of aromatic
stabilization. The last three examples are compounds in which the charge is
delocalized by resonance (protonation occurs at the light blue nitrogen) or
internal hydrogen bonding (the proton sponge).
In contrast to the earlier examples of acid pKas, the values for
these ammonium cations are nearly identical in water and DMSO solvents.
Indeed, the fact that most of the DMSO pKas are a bit lower than
their water counterparts suggests that DMSO is slightly more basic than
water. In acetonitrile all the pKa values are about 10 units
higher than the DMSO values.
pKas for Some Ammonium Species in Different Solvents
|
 |
|---|
The influence of solvent on acidity and basicity noted above may cause unexpected changes in simple acid-base equilibria. If acetic acid and triethyl amine are mixed together in water, a rapid proton transfer takes place to give nearly quantitative formation of triethyl ammonium acetate. The conjugate acids differ in strength by 106 and the weakest is favored at equilibrium. When the same two compounds are combined in DMSO solution there is negligible proton transfer, since acetic acid is now over 103 times weaker an acid than the triethyl ammonium cation.
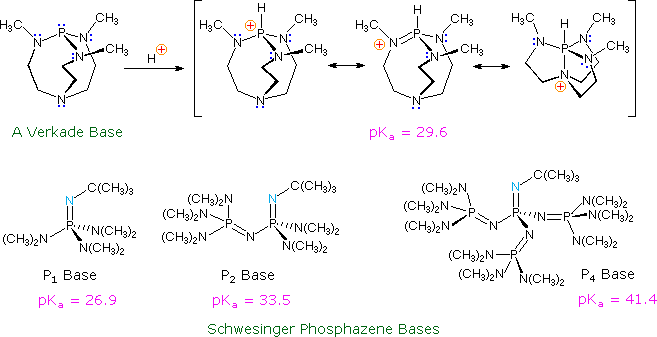
Nonionic Superbases
The strongest bases available to organic chemists are alkali metal
alkoxide salts, such as potassium tert.-butoxide, alkali metal alkyls, such
as n-butyl lithium, and amide salts of alkali metals, such as LDA. These
powerful bases are all potential nucleophiles (some more than others) and
have partially ionic bonds to the metal. Recently, a new class of
non-metallic, poorly nucleophilic, neutral bases have been prepared and
studied. Some examples are shown in the following diagram.
The basic site in the Verkade base is the phosphorous atom, the conjugate
acid being stabilized by transannular bonding to nitrogen. The strength of
these bases may be modified by substituents on the flanking nitrogens. The
Schwesinger phosphazene bases increase their strength as additional
phosphazene units are added in conjugation with the basic site (the light
blue nitrogen atom). All the pKas for these bases are measured
in acetonitrile.
3. Hybridization
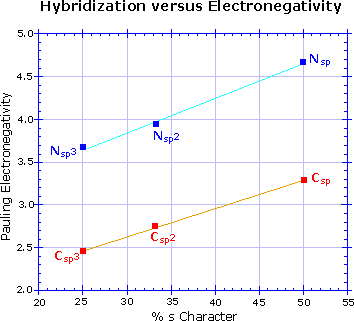
Hybridization has a strong influence on acidity, as shown by the three
carbon acids on the upper left below. The greater the s-character of the
orbital holding the electron pair of the conjugate base, the greater will
be the stability of the base. This corresponds to the lower energy of an
s-orbital compared with p-orbitals in the same valence shell. It also
corresponds to the increased electronegativity or inductive electron
withdrawal that is found for different hybridization states of a given
atom, as depicted in the graph on the right. The difference in acidity of
2-butynoic acid and butanoic acid, shown in the shaded box at lower left,
provides a further illustration of this inductive effect.
Carbocation stability is also influenced by hybridization, but in the
opposite direction (sp3 > sp2 > sp).
Carbon Acids
Inductive Effect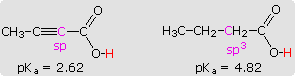
|
 |
|||||||||
4. Stereoelectronic Control of Enolization
Many carbon acids have enhanced acidity because of a neighboring
functional group. The acidity of alpha hydrogens in aldehydes,
ketones and esters
is well documented, and is the source of many important synthetic
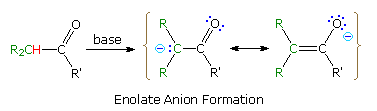
procedures. The following equation illustrates the general enolate anion
transformation, with the acidic alpha-hydrogen colored red. The resulting
ambident
anion is stabilized by charge delocalization, and may react with
electrophiles at both carbon and oxygen.
Stereoelectronic factors govern the enolization reaction, as illustrated by
clicking on the diagram below. The bond from the
alpha carbon to the acidic alpha-hydrogen must be oriented 90º to the plane
of the carbonyl group, or parallel to the pi-electron system (colored
magenta here). The ideal overlap occurs with a 0º dihedral angle between
this bond and the pi-orbital, as shown.
By clicking on the diagram a second time, the importance of this stereoelectronic requirement will be demonstrated. An increase in the acidity of carbon acids activated by two carbonyl groups is well known, and is illustrated by the two beta-dicarbonyl compounds on the left side of the diagram. In such cases the acidic C-H unit may be oriented perpendicular to both carbonyl groups, and the resulting planar anion is stabilized by additional charge delocalization (over both oxygens and the central carbon). In the case of the bicyclic diketone on the right, the C-H bond nearly eclipses the two carbonyl C-O bonds, resulting in a dihedral angle with the pi-electron systems of roughly 90º. Consequently, the acidity of this hydrogen is similar to that of the hydrogens of an alkane or cycloalkane. It should also be apparent that if an enolate anion were to be formed to the bridgehead carbon, the double bond would be prohibited by Bredt's rule.
5. Kinetic Acidity
The most common acid-base terminology, pKa , reflects an equilibrium acidity, extrapolated or normalized to water. In the following equation a base, B:(–) M(+), abstracts a proton from an acid, H-A, to form a conjugate acid - base pair (A:(–) M(+) & B-H). The rate of the forward proton abstraction is k f , and the reverse rate of proton transfer is k r. This kind of equilibrium is usually characterized by an equilibrium constant, Keq, which is the ratio of the rate constants (k f / k r). If H-A is a weaker acid than H-B the equilibrium will lie to the left, and Keq will be smaller than 1.
| H-A + B:(–) M(+) | 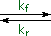 |
A:(–) M(+) + B-H |
| (acid1)(base1) | (base2)(acid2) |
In cases where H-A is very much weaker than H-B, Keq may be too small to measure, but it may be possible to determine the rate of the forward proton abstraction under certain circumstances. If an isotopically labeled conjugate acid of the base is used as a solvent for the reaction (B-D in the following equations), then any proton abstraction that occurs will be marked by conversion of H-A to D-A. The green shaded top equation shows the initial loss of the proton, and the second equation describes the rapid deuteration of the intermediate conjugate base, A:(–). As these reactions proceed, the H-A reactant will be increasingly labeled as D-A, and the rate of isotope exchange will indicate the kinetic acidity of H-A. It is assumed that kinetic acidity is roughly proportional to equilibrium (thermodynamic) acidity, but this is not always true.
| solvent = B-D | ||
| H-A + B:(–) M(+) | |
A:(–) M(+) + B-H |
| D-A + B:(–) M(+) | |
A:(–) M(+) + B-D |
The following diagram provides an instructive example of these
principles. The first equation, in the yellow shaded box, provides
important information about heavy water (deuterium oxide), which will be
used as a solvent for our experiment. Heavy water is similar to water in
many respects, but is 10% more dense and a ten-fold weaker acid. A 1 molar
concentration of sodium deuteroxide will serve as the base, and an
equimolar quantity of 3,3-dimethyl-1-butyne will serve as the weak acid.
The most acidic hydrogen in this hydrocarbon (colored red) is at C-1. In
practice, we would need to use a co-solvent to completely dissolve the
hydrocarbon in the heavy water, but this has been omitted in order to
simplify the discussion.
The second equation describes the essential changes expected on combining
these reactants in the heavy water solvent. Since the terminal alkyne is a
much weaker acid than heavy water, acid-base equilibria do not favor its
conjugate base. Nevertheless, if the acetylide anion is formed, even in low
concentration, it should react quickly by abstracting a deuterium from a
neighboring deuterium oxide molecule. The result would be an observable
exchange of deuterium for hydrogen, testifying that an acid-base reaction
has occurred.

The green shaded box contains equations that help us to interpret the
experimental results. In order to evaluate the equilibrium acidity of the
substrate, we would need to measure the equilibrium constant Keq
for the initial acid-base equilibrium, shown at the top of the shaded box.
Since we know the Ka 's of 3,3-dimethyl-1-butyne and heavy
water, we can estimate Keq by dividing the former (10
-25) by the latter (10 -17). This calculation reveals
a Keq that would be difficult to measure directly because of its
small magnitude (10 -8). Indeed, the equilibrium concentration
of acetylide anion is estimated to be only 2*!0 -10 M.
If we examine this experiment from the viewpoint of kinetics,
easily observable evidence of terminal alkyne acidity is obtained. The last
three rows of equations in the green shaded box make this clear. Since
Keq is the ratio of forward and reverse rate constants, it is
possible to draw conclusions about the rate of terminal proton abstraction
from the alkyne. This leads to the conclusion that reasonably rapid
hydrogen-deuterium exchange will occur, even though the acetylide anion is
never present in concentrations exceeding 10 -9 M.
This example also demonstrates the limits of the isotope exchange
approach. The 3,3-dimethyl-1-butyne substrate also has nine other hydrogen
atoms (colored orange) that do not exchange with deuterium under these
conditions. We know that these hydrogens are much less acidic
(Ka ca. 10 -48), and it is interesting to consider
their potential participation in acid-base reactions by the previous
analysis. The estimated Keq for such carbanion formation is
ca.10 -30, taking into account the nine-fold increase in
concentration. This implies a concentration of one carbanion in every
109 liters of solution. The kinetic analysis is equally
discouraging. The forward rate constant is estimated to be 10
-20 Ms-1. The time required to exchange half these
hydrogens for deuterium would therefore be about 1010
centuries!
In order to study the kinetic acidity of extremely weak acids
(pKa's = 30 to 50) it is necessary to use much stronger bases,
which of course have much weaker conjugate acids. Amide anions
(pKa's = 26 to 36) have been used for this purpose.
By comparing the rates of hydrogen exchange for different compounds under identical conditions, tables of relative kinetic acidities may be assembled. An interesting example of such a study has been reported for a group of nitroalkanes having acidic α-hydrogens. Compared with the terminal alkyne discussed above, such nitroalkanes are relatively strong C-H acids. Removal of an α-hydrogen by a base generates a conjugate base called an aci-anion, as shown here.
| R2CH-NO2 + B:(–) M(+) | |
R2C=NO2(–) M(+) + B-H |
| nitro compound | aci-conjugate base |
| Compound | pKa | Relative Rate of Exchange |
|---|---|---|
| CH3NO2 | 10.2 | 120 |
| CH3CH2NO2 | 8.5 | 20 |
| (CH3)2CHNO2 | 7.7 | 1.0 |
Since the nitroalkanes used in this study are stronger acids than water, the kinetic exchange experiments must be conducted under milder conditions than those used for the terminal alkyne. This is achieved by using smaller base concentrations and lowering the temperature of the exchange reaction. Accurate pKa's of 2-nitropropane, nitroethane and nitromethane may be measured directly in aqueous solution. These kinetic and equilibrium acidities are listed in the table on the right. Note that for these three compounds, kinetic acidity changes in an opposite fashion to equilibrium acidity. The kinetic order seems to reflect steric hindrance and carbanion stability; whereas, the equilibria favor increased substitution of the aci-anion double bond.
Base-catalyzed isotope exchange studies of compounds incorporating more
than one set of acidic hydrogens provides additional insight concerning the
creation and use of nucleophilic conjugate bases. Ketones provide many
examples of regioisomeric enolate base formation, and the following diagram
shows two such cases. As noted in the nitroalkane study, hydrogens on an
α-methyl group are exchanged more rapidly than those on more substituted
α-carbon atoms. The equations in the diagram show only the initial product
from a single exchange. These products have additional α-hydrogens which
are also exchanged by subsequent reactions of this kind, so that complete
replacement of all α-hydrogens by deuterium takes place in a short
time.
The relative stability of the resulting enolates increases with
substitution of the enolate double bond. Equations showing the equilibrium
concentrations of these isomeric enolates will be displayed by clicking the
Toggle Equations button. In order to
determine enolate anion equilibria for these ketones, the bulky strong base
sodium hexamethyldisilazide (pKa = 26) was used.
By clicking the Toggle Equations button a second time, the relative rates of α-hydrogen exchange for some substituted cyclohexanones will be displayed above. Once again, less substituted α-carbons exchange more rapidly, but more highly substituted enolates are found to predominate under equilibrium conditions. A third click of the Toggle Equations button will display an energy profile for the 2-methylcyclohexanone case, which should clarify the distinction between kinetic and equilibrium acidity. Two other examples are also shown. These displays may be cycled repeatedly.
Most carbon acids yield conjugate bases that are stabilized by charge delocalization onto neighboring heteroatoms. This resonance stabilization requires significant structural reorganization of the initial compound, which in turn imposes an energy barrier that retards the rate of proton abstraction. For example, the alpha-carbon of a ketone or ester must undergo rehybridization as the enolate anion is formed. The stereoelectronic demands of this change were described above, and it is not surprising that enolate anion formation is much slower than equivalent proton transfers between alcohols and other hydroxylic compounds. Deprotonation rates of phenol and nitromethane, compounds with nearly identical pKa's (10.0), provide an instructive example of this structural reorganization factor. The acidic proton in phenol is bound to oxygen, so deprotonation requires little structure change and is very fast. Nitromethane is a carbon acid. Deprotonation to an aci-anion involves considerable structural change, and is a million times slower than phenolate formation. These structural changes are illustrated in the following diagram.
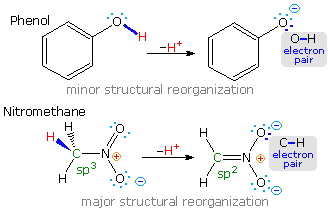
Note that the O-H electron pair in phenol remains largely on oxygen in the corresponding conjugate base, whereas the C-H electron pair in nitromethane is predominantly shifted to oxygen in its conjugate base (colored blue).
The trends outlined here are a bit oversimplified, since solvent and cation influences have been ignored. For a discussion of these factors, and practical applications of enolate anion intermediates in synthesis Click Here.
Substituent Effects on the Acidity of Carboxylic Acids
The carboxylic acids are a large and structurally diverse class of compounds. Since most are at least partially soluble in water and have pKa's in the 2 to 5 region, the influence of functional substituents and structural features on aqueous acidity have been studied extensively. Formic acid, HCO2H, is the simplest member of this class, and will serve as a useful reference point, pKa=3.75. Although the greater acidity of formic acid compared with methanol has been attributed to resonance stabilization of the formate anion, the different solvation demands of the respective conjugate anions result in an entropy difference that also favors the formate base. Both factors are depicted in the following illustration.. Resonance delocalization of the negative charge in the formate anion produces a large enthalpic stabilization shown by the magenta arrow. In water solution both methanol and formic acid are incorporated into the dynamic hydrogen bonded structure of liquid water. On ionization, each of these solutes produces a hydrated proton (hydronium ion) and a negatively charged conjugate base. The hydronium ion is common to both cases and can be ignored. The negative charge in the methoxide anion is concentrated on a single oxygen atom and demands strong solvation by water molecules, indicated by the aqua-colored dots. This solvation forces significant structural organization on many water molecules at the cost of decreased entropy. The formate anion also carries a single negative charge, but it is distributed over two oxygen atoms, so the charge density at either site is halved, compared with methoxide. This lower charge density demands much less solvation by water, resulting in a smaller entropy cost.
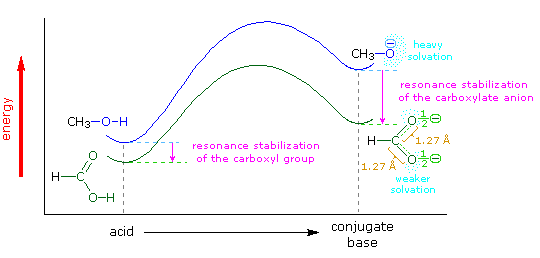
The importance of solvation and the accompanying entropy changes to any discussion of acidity may be seen by comparing the pKa's of methanol and formic acid in water and DMSO, a solvent that poorly solvates anions. In water the pKa of methanol is 15.5, nearly 12 powers of ten less acidic than formic acid (3.75). In DMSO the pKa's of methanol and formic acid are roughly 29 and 13 respectively, representing a very large decrease in Brønsted acid strength for both compounds (more than ten powers of ten). Furthermore, the difference in acid strength between methanol and formic acid in DMSO is magnified about ten thousand times, even though the enthalpic resonance stabilization presumably remains constant. A more extensive discussion of solvent effects on acidity was presented earlier. When comparing the acidities of different acids, care must be taken to use pKa's measured in the same solvent. In this discussion all the pKa's were taken in or extrapolated to water at 25 ºC. Measurements in mixed aqueous solvents, using water-soluble organic co-solvents such as ethanol, acetonitrile, dioxane, DMSO and acetone, generally give significantly larger pKa's.
In all other carboxylic acids an organic substituent replaces the hydrogen of formic acid, and it is instructive to analyze the change in acid strength caused by this change. To begin with, we must recognize that the carbonyl moiety of the carboxyl group is electrophilic and withdraws electrons from substituents. The deactivating nature of the carboxyl group on electrophilic substitution of benzoic acid is one example of this property. Resonance structures, such as A, B & C in the following diagram, are often drawn to describe this electrophilic character. The inductive effect of substituent Z in this diagram may enhance or diminish this character, depending on its overall electronegativity. Inductive electron withdrawal will increase the electrophilic character and the acidity of the carboxyl group, as shown in the green shaded box on the right. Resonance electron donation, either by p-π or π-π interaction, would act to stabilize the carboxylic acid, reducing its electrophilicity and acidity. These two effects often act in opposition, and in the case of carbonic acid ( H2CO3 ) electron donation overcomes inductive withdrawal, resulting in a pKa1=6.63.
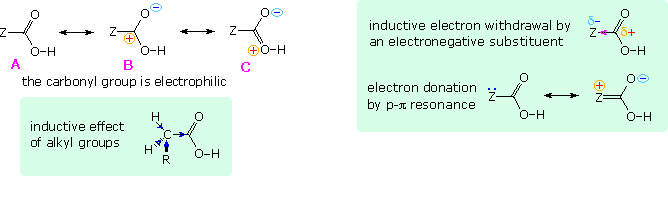
Saturated aliphatic acids are generally ten times weaker than formic
acid, which may seem surprising since carbon has a higher Pauling
electronegativity than hydrogen (2.55 versus 2.20). However, we must
recognize that a carbon atom is larger and more polarizable than hydrogen,
allowing it to shift electrons toward the more electronegative carbonyl
carbon of the carboxyl group. Also, hydrogen and alkyl substituents on the
α-carbon assist in this inductive electron shift, as shown in the green box
on the left. This analysis is supported by the activating influence of
alkyl substituents in electrophilic aromatic substitution, the Markovnikov
rule, and the greater reactivity of aldehydes with nucleophiles
compared with equivalent methyl ketones.
The four carboxylic acids in the first row of the following table
illustrate the electron donating quality of alkyl groups. As the number of
carbon atoms in the group increases from one to five, the inductive
electron donation also increases. The compounds in the next three rows of
the table demonstrate that electronegative substituents on an alkyl group
can shift its inductive effect from donating to withdrawing (relative to
hydrogen). Thus, all the haloacetic acids are more acidic than formic acid,
with fluoroacetic acid being the most acidic. Additional halogen
substituents have an additive influence, and moving the substituent from
the α to a β-carbon reduces its influence on the acidity. Note that a
hydroxyl substituent has a much weaker effect than any of the halogens,
despite the higher electronegativity of oxygen (3.44 compared with 3.16 for
chlorine).
| Compound | pKa | Compound | pKa | Compound | pKa | Compound | pKa | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CH3CO2H | 4.76 | CH3CH2CO2H | 4.87 | CH3(CH2)2CO2H | 4.91 | (CH3)3CCO2H | 5.05 | |||
| FCH2CO2H | 2.59 | ClCH2CO2H | 2.85 | BrCH2CO2H | 2.89 | ICH2CO2H | 3.13 | |||
| NCCH2CO2H | 2.50 | HOCH2CO2H | 3.82 | Cl2CHCO2H | 1.25 | Cl3CCO2H | 0.77 | |||
| NCCH2CH2CO2H | 3.98 | ClCH2CH2CO2H | 3.95 | BrCH2CH2CO2H | 4.00 | ICH2CH2CO2H | 4.06 |
Conjugation and Hybridization
The aliphatic acids discussed above do not provide any insight into p-π or π-π conjugation effects, since the sp3-hybridized α-carbon insulates the carboxyl group from such interactions. Conjugation may be studied by using α,β-unsaturated and aromatic carboxylic acids. The two parent compounds of these classes, acrylic acid (CH2=CHCO2H) and benzoic acid (C6H5CO2H), are both slightly stronger than acetic acid and have similar pKa's of 4.26 and 4.20 respectively. Since their influence is probably a combination of inductive and resonance effects, it would be helpful to evaluate one of these alone. The following four compounds represent acetic acid derivatives in which a methyl hydrogen has been replaced with a methyl group, a vinyl group, a phenyl group and a chlorine atom respectively. In each compound a methylene group insulates the substituent from the carboxyl group, prohibiting conjugative interactions. As noted above, the methyl substituent is weakly electron donating and the chlorine exerts a strong electron withdrawing influence. Comparatively, the vinyl and phenyl groups have an electron withdrawing inductive effect roughly 25% that of chlorine. From this we may conclude that resonance electron donation to the carboxyl function in acrylic acid and benzoic acid substantially dilutes the inductive effect of the sp2 substituent groups.
|
The increased electronegativity of sp2 and sp hybridized
carbon compared with sp3 carbon was noted
earlier. This increase is particularly dramatic for triply bonded
substituents, as seen in the acidity of 2-propynoic acid,
HC≡CCO2H, and 3-butynoic acid,
HC≡CCH2CO2H, having respective pKa's of
1.90 and 3.30. Conjugative electron donation in 2-propynoic acid is very
small, compared with acrylic acid, reflecting the poor electron donating
character of the triple bond.
Another aspect of conjugation concerns the ability of a double bond, triple
bond or aromatic ring to transmit the influence of a remote substituent to
the carboxyl group. The compounds in the following table provide
information bearing on this issue. The top row consists of β-substituted
acrylic acid derivatives. Methyl and phenyl substituents exert a weakening
effect; whereas chlorine strengthens the acid. Comparing these
relationships with similar substituent effects in equivalent saturated
acids (previous table) leads to some interesting
differences.
• A β-chlorine substituent exerts the same acidity strengthening effect
regardless of unsaturation in the connecting chain.
• A β-methyl group decreases the acidity of the unsaturated acid ten fold
over that of the saturated analog.
• A β-phenyl group increases the acidity of the saturated acid, but
decreases that of the unsaturated acid by roughly the same degree.
These observations may be interpreted in several ways. First, the inductive
electron withdrawal by chlorine through a C–C sigma-bond is about the same
as through a pi-bond. Second, The inductive electron donation by a methyl
group occurs to a significant degree by hyperconjugation
or conjugated hyperconjugation. Finally, the curious inversion of the
phenyl influence may be attributed to an exclusive inductive electron
withdrawal down the saturated connecting group, overpowered by a
conjugative donation through the unsaturated chain.
| Compound | pKa | Compound | pKa | Compound | pKa | Compound | pKa | |||
|---|---|---|---|---|---|---|---|---|---|---|
| t-CH3CH=CHCO2H | 4.74 | (CH3)2C=CHCO2H | 5.12 | ClCH=CHCO2H | 3.32 | t-C6H5CH=CHCO2H | 4.50 | |||
| p-CH3C6H4CO2H | 4.36 | p-ClC6H4CO2H | 3.98 | p-CH3OC6H4CO2H | 4.48 | p-O2NC6H4CO2H | 3.42 | |||
| m-CH3C6H4CO2H | 4.27 | m-ClC6H4CO2H | 3.82 | m-CH3OC6H4CO2H | 4.10 | m-O2NC6H4CO2H | 3.47 |
The substituted benzoic acids in the above table exhibit many of the
same effects noted for the acrylic acid derivatives. It must, however, be
noted that the meta and para-substituent locations in these compounds are
further removed from the carboxyl group, both in distance and number of
connecting bonds, than in the acrylic acid examples. This will reduce the
magnitude of any inductive effects. The para-location permits conjugative
interaction of the substituent with the carboxyl function; the meta
location does not. For comparison purposes remember that benzoic acid
itself has a pKa = 4.2.
A para-methyl substituent appears to have double the electron donating
effect of a meta-methyl group, again suggesting that conjugative
hyperconjugation may be important. The meta-chlorobenzoic acid isomer is
significantly more acidic than the para-isomer, largely because it is
closer to the carboxyl function, and in part due to resonance electron
donation by the para-chlorine. The two methoxybenzoic acids are
particularly informative, inasmuch as the meta isomer has a slightly
increased acidity, whereas the para-isomer is significantly weakened.
Oxygen has a much larger electronegativity than carbon, but it is an
excellent p-π electron donor to sp2 carbon functions. For the
meta isomer, the inductive effect is somewhat stronger than the resonance
donation, but the para-isomer is able to donate an oxygen electron pair
directly into the electrophilic carboxyl function. Both the meta and
para-nitro substituent withdraw electrons from the benzene ring by a
combination of inductive and resonance action, and the corresponding acids
are greatly strengthened. A quantitative treatment of meta and
para-substituent effects on the properties and reactions of benzoyl
derivatives has been developed by L.P.
Hammett.
The Ortho Effect
In general, ortho-substituted benzoic acids are stronger acids than
their meta and para isomers, regardless of the nature of the substituent.
The ortho effect is large for the nitrobenzoic acids, which show nearly a
20 fold increase in acidity, roughly an 8 fold factor for the halobenzoic
acids, and a 2.5 to 3 fold increase for methyl and cyano substituents. The
methoxybenzoic acids are exceptional, in that the ortho and meta isomers
have nearly identical pKa's (ca. 4.1), presumably due to the
exceptional p-π electron donation from oxygen noted above.
Many of the factors that influence the acidity of substituted benzoic acids
are summarized in the following diagram. First, although the phenyl group
is inductively electron withdrawing, it can donate electrons to a carboxyl
group by π-π resonance, as shown in the green shaded box in the upper left.
A substituent Y may perturb the balance of these two factors by its
inductive influence or by resonance. Two resonance cases, one showing
electron withdrawal by a nitro substituent and the other electron donation
by a methoxy substituent, are shown to the right of the green box.
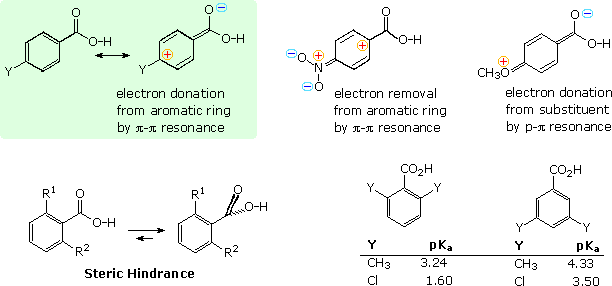
The increased acidity of ortho-substituted benzoic acids is attributed
to steric hindrance that forces the carboxyl group to twist out of the
plane of the benzene ring. The inductive character of the phenyl group does
not change with such twisting, but resonance ( conjugative electron
donation ) requires a coplanar relationship. For example, ortho-toluic acid
( R1 = CH3 & R2 = H ) has a
pKa of 3.9 compared with 4.2 for benzoic acid itself. If the
methyl is changed to a larger tert-butyl group the pKa
drops to 3. 53. By sandwiching the carboxyl group between two ortho
substituents, it is forced to lie perpendicular to the plane of the
aromatic ring, and conjugation is prohibited completely. The dimethyl and
dichlorobenzoic acid isomers shown at the lower right in the diagram
provide dramatic evidence of this conformational effect, with the bis-ortho
(2,6-) isomers representing the exclusive action of the inductive
effect.
Steric interference with conjugation may also perturb the acidity of
acyclic unsaturated acids. Thus, 2,3-dimethyl-2-butenoic acid has a
pKa of 4.41, compared with the 5.12 pKa of
3-methyl-2-butenoic acid.
Hydrogen Bonding
The presence of a hydrogen bond donor near a carboxyl group may act to enhance its acidity, as demonstrated by the three isomeric hydroxybenzoic acids and three isomeric benzenedicarboxylic acids (phthalic acids) shown in the following table. Compared with benzoic acid, the meta and para-isomers display expected changes in acidity, due to combined inductive and resonance effects. However, the ortho isomers are both roughly 15 times more acidic, even though the hydroxyl and carboxyl substituents have opposite influences in the para-location.
| ortho relationship | pKa1 | pKa2 | meta relationship | pKa1 | pKa2 | para relationship | pKa1 | pKa2 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| salicylic acid | 2.97 | 13.44 | meta-hydroxybenzoic acid | 4.08 | 9.91 | para-hydroxybenzoic acid | 4.58 | 9.40 | ||
| phthalic acid | 2.98 | 5.28 | isophthalic acid | 3.46 | 4.46 | terephthalic acid | 3.51 | 4.82 |
Intramolecular hydrogen bonding of an ortho OH donor to the carbonyl oxygen of the carboxyl group, acting as an acceptor, increases the positive charge on the carbonyl carbon and consequently the acidity of the carboxyl OH. This is illustrated for salicylic acid in the following diagram. Phthalic acid engages in a similar seven-membered cyclic hydrogen bond with a similar outcome. This intramolecular hydrogen bonding also explains the decreased acidity of the remaining acidic function - that is the phenolic OH in salicylic acid and the second carboxyl group in phthalic acid.
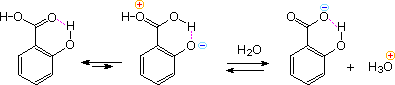
The stereoisomeric 2-butenedioic acids, maleic and fumaric acid display a similar behavior. The cis isomer, maleic acid, has pKa1 = 2.00 and pKa2 = 6.50. This contrasts with the values for the trans-isomer, fumaric acid, pKa1 = 3.00 and pKa2 = 4.50.