Reduction Reagents
Reducing Reagents
| Reagent | Preferred Solvents | Functions Reduced | Reaction Work-up |
|---|---|---|---|
| Sodium Borohydride NaBH4 |
ethanol; aqueous ethanol 15% NaOH; diglyme avoid strong acids |
aldehydes to 1º-alcohols ketones to 2º-alcohols 1,2-reduction of enones is favored by CeCl3 inert to most other functions |
1) simple neutralization 2) extraction of product |
| Lithium Aluminum Hydride (LAH) LiAlH4 |
ether; THF avoid alcohols and amines avoid halogenated compounds avoid strong acids |
aldehydes to 1º-alcohols ketones to 2º-alcohols carboxylic acids to 1º-alcohols esters to alcohols epoxides to alcohols nitriles & amides to amines halides & tosylates to alkanes most functions react |
1) careful addition of water 2) dissolve aluminum salts 3) extraction of product |
| Lithium tri t-Butoxyaluminohydride LiAlH(Ot-C4H9)3 |
ether; THF avoid alcohols and amines avoid halogenated compounds avoid strong acids |
fast: acid chlorides to aldehydes (at -78 ºC) 3º-amides to aldehydes (at -78 ºC) nitriles to aldehydes (at -78 ºC) slower: aldehydes to 1º-alcohols ketones to 2º-alcohols |
1) careful addition of water 2) dissolve aluminum salts 3) extraction of product |
| Diisobutylaluminum Hydride AlH[CH2CH(CH3)2]2 |
THF; toluene avoid alcohols and amines avoid halogenated compounds avoid strong acids |
fast: acid chlorides to aldehydes (at -78 ºC) 3º-amides to aldehydes (at -78 ºC) nitriles to aldehydes (at -78 ºC) slower: aldehydes to 1º-alcohols ketones to 2º-alcohols |
1) careful addition of water 2) dissolve aluminum salts 3) extraction of product |
| Diborane B2H6 = 2 BH3 |
ether; THF sulfide complex in CH2Cl2 complexes with amines avoid alkenes & alkynes |
carboxylic acids to 1º-alcohols aldehydes to 1º-alcohols ketones to 2º-alcohols nitriles to amines esters & epoxides slowly reduced |
1) dilute acid or H2O2 2) extraction of product |
|
Hydrogen & Catalyst H2 & Pt, or Pd, or Ru, or Ni Modified (poisoned) Catalyst |
alcohols, ethers, hydrocarbons or carboxylic acids |
alkenes & alkynes to alkanes (fast) nitro groups to amines (fast) imines to amines (fast) aldehydes & ketones to alcohols (slow) nitriles to amines (slow) may remove benzylic groups alkynes to alkenes acyl chlorides to aldehydes |
filter to remove catalyst |
|
Reactive Metals Na, or Li, or K Mg or Al or Zn or Fe |
liq. ammonia & ether co-solvents or alcohols or amines water, alcohols, acetic acid or aqueous mineral acid |
ketones to 2º-alcohols alkynes to alkenes conjugated π-systems (e.g. aromatic rings, dienes & enones) cleaves C-X and benzylic groups cleaves activated substituents nitro groups to amines C=O (aldehyde/ketone) to CH2 |
1) quench with NH4Cl 2) extract product extract product from salts |
Many related hydride reagents, having different reductive power and selectivity, incorporate a variety of organic ligands on the hydride carrier atom. Among these are: lithium triethylborohydride (super hydride), potassium tri-sec-butylborohydride (K-Selectride), sodium bis(2-methoxyethoxy)aluminumhydride (Red-Al), sodium cyanoborohydride, sodium triacetoxyborohydride. various organosilanes and in situ generated aluminum hydride and dichlorohydride. Organoborane derivatives having modified reactivity relative to borane itself include: bis(1,2-dimethylpropyl)borane (disiamylborane), 9-borabicyclo[3.3.1]nonane (9-BBN) & catecholborane (BHcat).
End of this supplementary topic
Oxidation Reagents
Oxidation Reagents
| Reagent | Preferred Solvents | Functions Oxidized | Reaction Work-up |
|---|---|---|---|
| Jones Reagent H2CrO4 |
aqueous sulfuric acid & acetone (avoid acid sensitive systems) |
1º-alcohols to carboxylic acids aldehydes to carboxylic acids 2º-alcohols to ketones avoid amines and sufides |
1) destroy excess reagent 2) extract product |
| Collins Reagent CrO3 • 2 C5H5N |
methylene chloride (CH2Cl2) |
1º-alcohols to aldehydes 2º-alcohols to ketones |
1) filter inorganic salts 2) wash with aqueous acid 3) remove CH2Cl2 solvent |
| Pyridinium Chlorochromate ClCrO3 • C5H5NH |
methylene chloride (CH2Cl2) |
1º-alcohols to aldehydes 2º-alcohols to ketones |
1) filter inorganic salts 2) wash with aqueous acid 3) remove CH2Cl2 solvent |
| Dimethyl Sulfoxide (CH3)2S=O & DCC or Ac2O or (CF3CO)2O or SO3 or (COCl)2 |
CH2Cl2 or ethers or DMSO | A Mild Procedure 1º-alcohols to aldehydes 2º-alcohols to ketones |
1) neutralize reactants 2) remove solvents |
|
Potassium Permanganate KMnO4 Osmium Tetroxide OsO4 |
water and aqueous solvent mixtures pyridine often used catalytically |
aldehydes to carboxylic acids 2º-alcohols to ketones alkenes to vicinal-diols (vic.-glycols) alkynes to carboxylic acids avoid amines and sufides alkenes to vicinal-diols (vic.-glycols) |
1) destroy excess reagent 2) extract product |
|
Periodic Acid HIO4 Lead Tetraacetate Pb(OCOCH3)4 |
water or aqueous mixtures benzene or acetic acid |
vic.-glycols to carbonyl compounds vic.-glycols to carbonyl compounds |
1) destroy excess reagent 2) extract product |
| Peracids CH3CO3H C6H5CO3H, etc. |
CH2Cl2 or ethers | alkenes to epoxides ketones to esters avoid amines and sufides |
1) destroy excess reagent 2) extract product |
| Ozone O3 |
CH2Cl2 or CHCl3 (sometimes alcohol) |
cleaves alkenes & alkynes avoid benzene derivatives amines and sufides |
1) destroy excess reagent & ozonides 2) extract product |
End of this supplementary topic
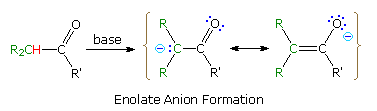
Alternatives to Enolate Anions
Reactions of Enolate-Like Species
1. Regioselectivity in Enolate Anion Formation and Reaction
The importance of enolate anions as synthetic intermediates is well
established. Nevertheless, problems remain concerning their selective
formation and reaction. For example, aldehyde enolate bases are likely to
undergo the aldol
reaction during their formation, and ketones like 2-heptanone have two
different alpha-carbons, each capable of enolization. The ambident
nature of enolate anions also enables electrophilic attack at both
oxygen and carbon, but in most synthesis applications bonding to carbon is
desired. Finally, enolate anions may often be formed as E/Z
stereoisomers, and it has been shown that reaction stereoselectivity,
when new chiral centers are created, depends on the enolate
configuration.
The following diagram illustrates how the conditions under which enolate
anion formation is accomplished can influence the regioselectivity
of the reaction. The two ketone substrates, 2-heptanone and
2-methylcyclohexanone, each have differently substituted alpha-carbons. In
each case, enolate anion mixtures are generated by reaction with a strong
2º-amide base (LDA is the
usual choice). If the ketone is added to a cold THF solution of excess
base, enolate anion formation is fast and irreversible (procedure
a). On the other hand, if a slight excess of ketone is allowed to
remain in solution, an equilibrium involving the ketone and the various
enolate species is established (procedure b). At equilibrium the
more stable enolate anion will predominate. The examples given in the
diagram also report results from an equilibrating preparation in which the
lithium metal in LDA is replaced by potassium (procedure c).
Regioselective Formation of Enolate Anions
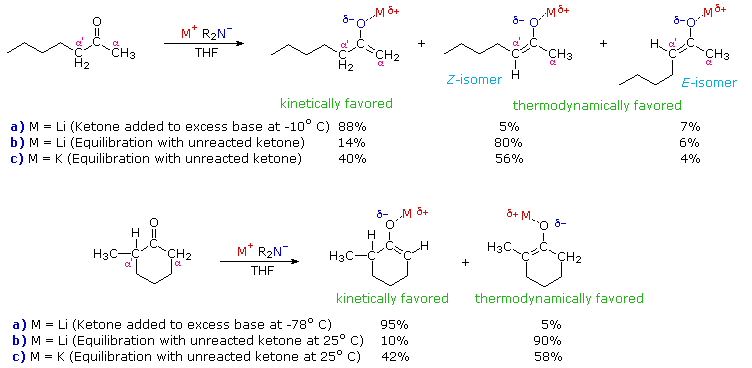
Several important principles are demonstrated here. First, if the enolate species has substantial double bond character, the more highly-substituted enolate double bond should predominate at equilibrium, as predicted from the stabilities of substituted alkenes. Since lithium-oxygen bonds are more covalent (have less ionic character) than potassium-oxygen bonds, the lithium enolate approximates an alkene more closely than the potassium enolate. Second, the greater ionic character of the potassium enolate places an increased negative charge on the alpha-carbon, a condition that is disfavored by alkyl group substitution. Indeed, the stability order of substituted carbanions is opposite to that of carbocations thanks to the electron donating character of alkyl groups relative to hydrogen. Finally, the rate of proton removal from an alpha-carbon site is decreased by alkyl substitution, probably reflecting a combination of steric hindrance (to bulky bases) and decreased carbanion stability. In both of the examples shown above, the conditions used in procedure (a) are typical of kinetically favored enolate formation, whereas those used in procedure (b) favor thermodynamic enolate formation. The comparative acidities provided by pKa values are derived from measurements made under equilibrating conditions, and therefore reflect thermodynamic acidity. Determinations of kinetic acidity require competitive isotope exchange experiments.
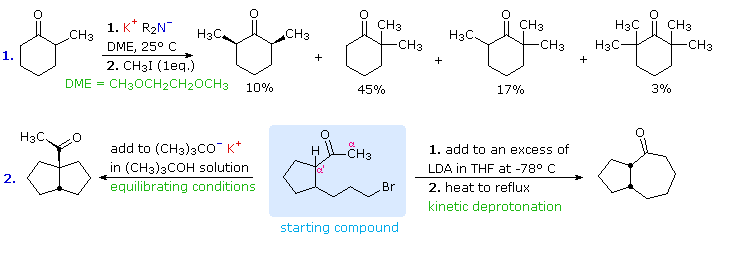
These principles influence the course of enolate alkylation reactions,
as shown in the following diagram. In the first case, 2-methylcyclohexanone
is converted to a thermodynamic enolate mixture, which is then reacted with
methyl iodide. The major product is the expected 2,2-dimethylcyclohexanone
(from the more stable enolate anion), but this is accompanied by di- and
trimethylated products together with about 20% unreacted starting material.
The complexity of the product mixture is due to acid-base proton transfer
between alkylated products and unreacted enolate anion. In other words,
once a small amount (say 5%) of dimethylcyclohexanone is formed, it finds
itself in solution with a relatively high concentration of a strong base
(the remaining enolate anion) that can remove another alpha-proton, giving
a new enolate anion that is further methylated. If the kinetically favored
lithium enolate (see the previous diagram) is used instead of the
equilibrium potassium enolates, 2,6-dimethylcyclohexanone is the chief
product.
The second reaction is an intramolecular alkylation that can occur in two
different ways. If the kinetically favored enolate (methyl proton removal)
is formed at low temperature, it reacts rapidly on warming to form a
seven-membered ring. Alternatively, the weaker base, potassium
tert-butoxide (in the alcohol as solvent), generates an equilibrium mixture
of enolates which eventually react by intramolecular alkylation. The
thermodynamically favored α'-enolate predominates, and the resulting
alkylation generates a five-membered ring.
Examples of Selective Enolate Alkylation
Another aspect of enolate anion alkylation, not yet addressed, is the
possibility of electrophilic bonding at oxygen. One example of such
behavior will be displayed by clicking the "Toggle Reactions"
button. Because of the substantial negative charge on the oxygen of
ambident anions, it might be expected that O-alkylation would be the rule
rather than the exception. This, in fact, is true when fully or extensively
ionized enolate salts are reacted with strong electrophiles. Ionization of
enolates is facilitated by high dielectric solvents, such as DMSO and DMF
(dimethylformamide), especially for potassium and cesium cation salts. As
shown in the lower part of the second diagram, the negatively charged
oxygens of DMSO cluster about a cation, providing substantial solvation
stabilization. No such solvation exists for the enolate anion, leaving it
open to reaction with an electrophile. Lithium enolates have significant
covalent character in the metal-oxygen bond, and this retards electrophile
attack at oxygen.
Ether solvents such as THF and DME (dimethoxyethane or glyme) are commonly
used for alkylations because they are inert to strong base and dissolve
enolate salts more effectively than hydrocarbons. The difunctional ether
DME (dimethoxyethane) is especially effective at solvating cations; and
this fact has led to the preparation of cyclic polyethers, known as
crown ethers, which are extraordinarily powerful solvating agents.
Crown ethers may be added to enolate salt solutions to enhance their
ionization. Indeed, the size of the crown ether can be tailored to fit the
cation being used, providing additional control over the course of enolate
reactions.
The nomenclature of crown ethers consists of two numbers. The first
(larger) number designates the overall ring size. The second number
indicates the number of ether oxygens. A symmetrical arrangement of the
oxygens in the ring is assumed.
2. Preparation and Reactions of Silyl Enol Ethers
One way of producing selective enolate anion intermediates is to first trap and isolate them as silyl enol ethers. These relatively stable compounds may then be used to generate isomerically pure enolate anions, or in some cases as enolic nucleophiles in their own right. In the following diagram, the first reaction illustrates the formation of a mixture of silyl enol ethers under equilibrating conditions. If a higher proportion of the minor isomer is desired the kinetically favored lithium enolate can be prepared and quenched with trimethylsilyl chloride. In either case the silyl ether mixture may be separated by distillation. Once a pure silyl ether isomer is in hand, it may be used to generate the corresponding lithium enolate in the manner shown. Alkylation reactions of these enolates then produces pure regioisomeric products.
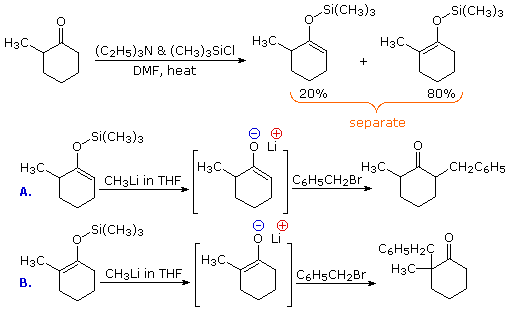
By clicking the "Toggle Reactions" button under the previous diagram, two examples of the direct use of silyl enol ethers will be displayed. Since the silyl ethers are not as reactive as enolate anions, the electrophiles with which they combine must be made more reactive. When carbonyl electrophiles are used, this can be accomplished by Lewis acid catalysts, as shown.
3. Enamines as Enolate Anion Surrogates
The formation of enamines by reaction of 2º-amines with aldehydes or
ketones has been
described. The double bond of the enamine transmits the nucleophilic
character of the nitrogen to the alpha-carbon, in a vinylagous
fashion. Because of the resulting ambident nucleophilicity of the enamines,
reactions with electrophiles may take place at either nitrogen or carbon.
Enamines derived from aldehydes are usually alkylated on nitrogen, an
undesirable course for most synthetic applications. Ketones give
significant C-alkylation, the thermodynamically favored course, as first
demonstrated by G. Stork (Columbia). The iminium ion
created by C-alkylation cannot react further, and is easily hydrolyzed to
the alkylated ketone. This is particularly useful if dialkylation products
are to be avoided. Thus, in the first example, direct methylation of the
enolate anion from this ketone gives significant amounts of the dimethyl
product, due to enolate proton exchange. As shown, the
enamine route gives only mono-methylated product.
The second example demonstrates that enamines may be acylated as well as
alkylated. In fact, the reversible nature of acylation removes the problem
of competing N-acylation. This case also illustrates the general tendency
to form the least substituted enamine when two different alpha-sites are
present. Conjugation of the non-bonding electron pair on nitrogen with the
pi-electrons of the double bond forces the alkyl substituents on nitrogen
to lie in the same plane as the double bond (see the resonance equation
displayed by the "Toggle Mechanism" button). As a result
substitution of the double bond leads to increased steric hindrance with
the nitrogen substituents. The five-membered cyclic 2º-amine pyrrolidine is
widely used for enamine reactions, in part because this steric hindrance is
minimized.
Examples of Enamine Reactions
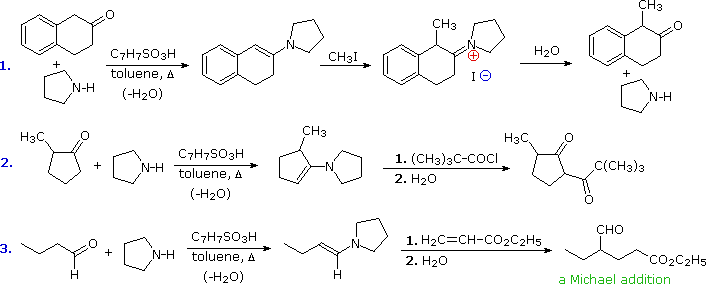
As noted, N-alkylation of enamines is common for aldehydes and some ketones. Michael addition reactions avoid this problem thanks to their reversibility. The third example shows such a reaction, and the "Toggle Mechanism" button displays a possible mechanism. The C-alkylation intermediate is thermodynamically more stable than the N-alkylation species, so it predominates at equilibrium. Both the charges in this intermediate are stabilized by delocalization, and hydrolysis rapidly converts it to the aldehyde-ester product. An interesting alternative is ring closure to a neutral enol ether compound (shown in the blue shaded box) which would also be hydrolyzed to the same product.
4. Imine and Hydrazone Anions
Still another way of circumventing some of the undesired aspects of enolate anion chemistry is to replace the oxygen of an aldehyde or ketone substrate with a 1º-amino group, in other words, to convert the carbonyl function to an imine. Imine derivatives are relatively easy to prepare, starting with an aldehyde or ketone and a 1º-amine or hydrazine derivative. The resulting C=N function does not activate alpha-C-H groups as effectively as a carbonyl function, but very strong bases such as LDA, alkyl lithiums and Grignard reagents will convert imines to their enamide conjugate bases quantitatively. This general reaction is shown in the green shaded box below.
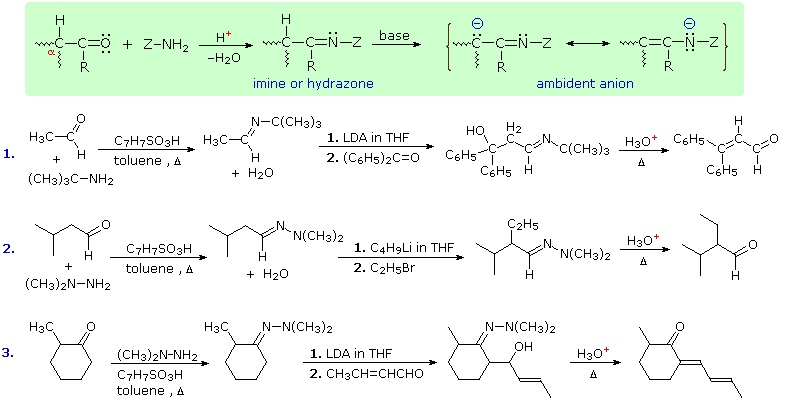
Three illustrations of the use of enamide bases in synthesis are displayed above. The first two examples use aldehyde derivatives, and if we were to attempt these reactions with the aldehyde enolate anion itself, aldol dimerization would result. The C=N function of imines is a poor acceptor of nucleophiles, so it does not assume such a role in aldol-like reactions. The third reaction is an aldol condensation in which a ketone serves as the donor. If cuprous salts are introduced before the unsaturated aldehyde is added to the enamide solution, conjugate addition takes place in preference to the 1,2-aldol addition.
End of this supplementary topic
Structure & Acidity
Molecular Structure and Acidity
1. Equilibrium Acidity
pKa Acidities of Some Common Hydrides
| 4 | 5 | 6 | 7 |
|---|---|---|---|
| CH3-H ca. 50 |
NH2-H 34 |
HO-H 15.74 |
F-H 3.2 |
| HS-H 6.97 (pK1) |
Cl-H -3 |
||
| HSe-H 3.8 (pK1) |
Br-H -6 |
||
| HTe-H 2.6 (pK1) |
I-H -7 |
The relative acidities of different acids are commonly measured and cited as pKa values, relative to a standard solvent base, often water. These numbers reflect the equilibrium acidities of the acids. An astounding range of acidities is displayed by even rather simple compounds. The table on the right lists some elemental hydrides from groups 4 through 7 of the periodic table. The pKa's determined (or in some cases estimated) for these compounds are shown beneath the formulas. Approximate values for higher members of group 4 and 5 hydrides (e.g. silane and phosphine) have not been reported. Note that these logarithmic numbers encompass nearly sixty powers of ten. This is a greater span than that encompassed by distance measurements starting from the radius of a hydrogen atom and extending to the diameter of the known universe.
Why do these relatively simple compounds differ in acid strength so
markedly? Two factors may be discerned:
• First, the compounds in the top row clearly show the importance of
electronegativity.
All the heavier elements have greater electronegativities than hydrogen,
with carbon being the least different. The ionic character of these
covalent bonds is such that hydrogen carries a partial positive charge, and
the heavier atom a corresponding negative charge. The greatest charge
separation is in H-F, where the electronegativity difference is nearly 2.
Removal of a proton is facilitated by this charge separation. The covalent
bond energies do not correlate inversely with acid strength, as one might
have expected, since the two strongest acids have the strongest bonds (H-O
111 kcal/mol & H-F 135 kcal/mol). Finally, the heavy atoms in the top
row have similar sizes, the covalent radii being 0.75 ±0.02 Å. The
importance of this fact will become apparent in the following
discussion.
• Second, the compounds in the columns representing periodic groups 6 and 7
show an increase in acidity moving from the top to the bottom. This is
opposite to the electronegativity change, and is best attributed to an
increase in heavy atom size. When an acid transfers a proton to a base, the
remaining residue (the conjugate base) must carry a negative charge.
Ignoring solvent stabilization (solvation),
the stability of ions is a function of charge density. A small ion has a
higher charge density than a larger ion of the same charge, making the
smaller ion less stable. From the covalent radius of oxygen compared with
sulfur, and fluorine compared with chlorine, it can be estimated that the
charge density on the larger atom is half that of the smaller. The
resulting stabilization of the conjugate base more than compensates for the
decrease in electronegativity in moving down the column; so H2S
is a stronger acid than H2O, and HCl a stronger acid than HF.
Since sulfur and chlorine are nearly the same size (covalent radii being
1.02 ±0.02 Å), electronegativity explains the difference in acidity between
H2S and HCl.
| NH4(+) 9.24 |
OH3(+) -1.74 |
S-H(–) 15 (pK2) |
Se-H(–) 11 (pK2) |
If the heavy atom of an acid carries a formal charge, its acidity will be changed substantially. This is demonstrated by the examples on the right. Ammonium and hydronium ions carry a positive charge, and the acidity of the species is increased by over fifteen powers of ten relative to uncharged ammonia and water. By contrast, hydrogen sulfide and hydrogen selenide are dibasic acids (they have two acidic protons). Once the first proton has been lost, the acidity of the negatively charged conjugate base is reduced over a million fold. This is true for most other dibasic acids such as H2SO4 and H2CO3.
Accurate acidity measurements in the pKa range from 1 to 14
can usually be made in water solution. However, acids stronger than the
hydronium ion (H3O(+)) and bases stronger than
hydroxide ion (OH(–)) react immediately with this solvent, and
the resulting "leveling effect" prevents direct measurement of their
pKa's. One way of circumventing this difficulty is to examine
the acidity of very strong ( pKa < 0) and very weak (
pKa > 15) acids in different (non-aqueous) solvents, and to
extrapolate these measurements to water. For example, solvents such as
acetic acid, acetonitrile and nitromethane are often used for studying very
strong acids. Very weakly acidic solvents such as DMSO, acetonitrile,
toluene, amines and ammonia are used to study the acidities of very weak
acids. The errors introduced in extreme cases, such as methane, are often
large; but the overall range of acid strengths observed in this manner
cannot be questioned.
It must be recognized that pKa values for the same compound
measured in different solvents will generally be different. The two most
common solvents in which such measurements have been made are water and
DMSO. The following table presents data for a few representative compounds,
with the pKa difference noted in the right hand column. Since
anion
solvation by water is superior to that provided by DMSO, the
pKa values from the latter solvent tend to be higher. However, a
decrease in charge density, as with sulfur, or internal delocalization of
negative charge, as in the last four compounds, lessens this
difference.
| Compound | H2O pKa | DMSO pKa | Δ pKa |
|---|---|---|---|
| H2O | 15.7 | 31.2 | 15.5 |
| H2S | 7.0 | 14.7 | 7.7 |
| C6H5OH | 10.0 | 18.0 | 8.0 |
| O2NCH3 | 10.2 | 17.2 | 7.0 |
| C6H5COCH3 | 18 | 24.7 | 6.7 |
| (CH3CO)2CH2 | 8.9 | 13.3 | 4.5 |
2. Hybridization
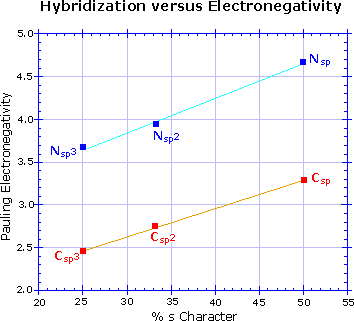
Hybridization has a strong influence on acidity, as shown by the three
carbon acids on the upper left below. The greater the s-character of the
orbital holding the electron pair of the conjugate base, the greater will
be the stability of the base. This corresponds to the lower energy of an
s-orbital compared with p-orbitals in the same valence shell. It also
corresponds to the increased electronegativity or inductive electron
withdrawal that is found for different hybridization states of a given
atom, as depicted in the graph on the right. The difference in acidity of
2-butynoic acid and butanoic acid, shown in the shaded box at lower left,
provides a further illustration of this inductive effect.
Carbocation stability is also influenced by hybridization, but in the
opposite direction (sp3 > sp2 > sp).
Carbon Acids
Inductive Effect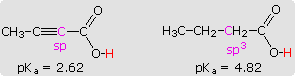
|
 |
|||||||||
3. Stereoelectronic Control of Enolization
Many carbon acids have enhanced acidity because of a neighboring
functional group. The acidity of alpha hydrogens in aldehydes,
ketones and esters
is well documented, and is the source of many important synthetic
procedures. The following equation illustrates the general enolate anion
transformation, with the acidic alpha-hydrogen colored red. The resulting
ambident
anion is stabilized by charge delocalization, and may react with
electrophiles at both carbon and oxygen.
Stereoelectronic factors govern the enolization reaction, as illustrated by
clicking on the diagram below. The bond from the
alpha carbon to the acidic alpha-hydrogen must be oriented 90º to the plane
of the carbonyl group, or parallel to the pi-electron system (colored
magenta here). The ideal overlap occurs with a 0º dihedral angle between
this bond and the pi-orbital, as shown.
By clicking on the diagram a second time, the importance of this stereoelectronic requirement will be demonstrated. An increase in the acidity of carbon acids activated by two carbonyl groups is well known, and is illustrated by the two beta-dicarbonyl compounds on the left side of the diagram. In such cases the acidic C-H unit may be oriented perpendicular to both carbonyl groups, and the resulting planar anion is stabilized by additional charge delocalization (over both oxygens and the central carbon). In the case of the bicyclic diketone on the right, the C-H bond nearly eclipses the two carbonyl C-O bonds, resulting in a dihedral angle with the pi-electron systems of roughly 90º. Consequently, the acidity of this hydrogen is similar to that of the hydrogens of an alkane or cycloalkane. It should also be apparent that if an enolate anion were to be formed to the bridgehead carbon, the double bond would be prohibited by Bredt's rule.
4. Kinetic Acidity
The most common acid-base terminology, pKa , reflects an equilibrium acidity, extrapolated or normalized to water. In the following equation a base, B:(–) M(+), abstracts a proton from an acid, H-A, to form a conjugate acid - base pair (A:(–) M(+) & B-H). The rate of the forward proton abstraction is k f , and the reverse rate of proton transfer is k r. This kind of equilibrium is usually characterized by an equilibrium constant, Keq, which is the ratio of the rate constants (k f / k r). If H-A is a weaker acid than H-B the equilibrium will lie to the left, and Keq will be smaller than 1.
| H-A + B:(–) M(+) | 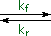 |
A:(–) M(+) + B-H |
| (acid1)(base1) | (base2)(acid2) |
In cases where H-A is very much weaker than H-B, Keq may be too small to measure, but it may be possible to determine the rate of the forward proton abstraction under certain circumstances. If an isotopically labeled conjugate acid of the base is used as a solvent for the reaction (B-D in the following equations), then any proton abstraction that occurs will be marked by conversion of H-A to D-A. The green shaded top equation shows the initial loss of the proton, and the second equation describes the rapid deuteration of the intermediate conjugate base, A:(–). As these reactions proceed, the H-A reactant will be increasingly labeled as D-A, and the rate of isotope exchange will indicate the kinetic acidity of H-A. It is assumed that kinetic acidity is roughly proportional to equilibrium (thermodynamic) acidity, but this is not always true.
| solvent = B-D | ||
| H-A + B:(–) M(+) |  |
A:(–) M(+) + B-H |
| D-A + B:(–) M(+) | |
A:(–) M(+) + B-D |
The following diagram provides an instructive example of these
principles. The first equation, in the yellow shaded box, provides
important information about heavy water (deuterium oxide), which will be
used as a solvent for our experiment. Heavy water is similar to water in
many respects, but is 10% more dense and a ten-fold weaker acid. A 1 molar
concentration of sodium deuteroxide will serve as the base, and an
equimolar quantity of 3,3-dimethyl-1-butyne will serve as the weak acid.
The most acidic hydrogen in this hydrocarbon (colored red) is at C-1. In
practice, we would need to use a co-solvent to completely dissolve the
hydrocarbon in the heavy water, but this has been omitted in order to
simplify the discussion.
The second equation describes the essential changes expected on combining
these reactants in the heavy water solvent. Since the terminal alkyne is a
much weaker acid than heavy water, acid-base equilibria do not favor its
conjugate base. Nevertheless, if the acetylide anion is formed, even in low
concentration, it should react quickly by abstracting a deuterium from a
neighboring deuterium oxide molecule. The result would be an observable
exchange of deuterium for hydrogen, testifying that an acid-base reaction
has occurred.

The green shaded box contains equations that help us to interpret the
experimental results. In order to evaluate the equilibrium acidity of the
substrate, we would need to measure the equilibrium constant Keq
for the initial acid-base equilibrium, shown at the top of the shaded box.
Since we know the Ka 's of 3,3-dimethyl-1-butyne and heavy
water, we can estimate Keq by dividing the former (10
-25) by the latter (10 -17). This calculation reveals
a Keq that would be difficult to measure directly because of its
small magnitude (10 -8). Indeed, the equilibrium concentration
of acetylide anion is estimated to be only 2*!0 -10 M.
If we examine this experiment from the viewpoint of kinetics,
easily observable evidence of terminal alkyne acidity is obtained. The last
three rows of equations in the green shaded box make this clear. Since
Keq is the ratio of forward and reverse rate constants, it is
possible to draw conclusions about the rate of terminal proton abstraction
from the alkyne. This leads to the conclusion that reasonably rapid
hydrogen-deuterium exchange will occur, even though the acetylide anion is
never present in concentrations exceeding 10 -9 M.
This example also demonstrates the limits of the isotope exchange
approach. The 3,3-dimethyl-1-butyne substrate also has nine other hydrogen
atoms (colored orange) that do not exchange with deuterium under these
conditions. We know that these hydrogens are much less acidic
(Ka ca. 10 -48), and it is interesting to consider
their potential participation in acid-base reactions by the previous
analysis. The estimated Keq for such carbanion formation is
ca.10 -30, taking into account the nine-fold increase in
concentration. This implies a concentration of one carbanion in every
109 liters of solution. The kinetic analysis is equally
discouraging. The forward rate constant is estimated to be 10
-20M s-1. The time required to exchange half these
hydrogens for deuterium would therefore be about 1010
centuries!
In order to study the kinetic acidity of extremely weak acids
(pKa's = 30 to 50) it is necessary to use much stronger bases,
which of course have much weaker conjugate acids. Amide anions
(pKa's = 26 to 36) have been used for this purpose.
By comparing the rates of hydrogen exchange for different compounds under identical conditions, tables of relative kinetic acidities may be assembled. An interesting example of such a study has been reported for a group of nitroalkanes having acidic α-hydrogens. Compared with the terminal alkyne discussed above, such nitroalkanes are relatively strong C-H acids. Removal of an α-hydrogen by a base generates a conjugate base called an aci-anion, as shown here.
| R2CH-NO2 + B:(–) M(+) |  |
R2C=NO2(–) M(+) + B-H |
| nitro compound | aci-conjugate base |
| Compound | pKa | Relative Rate of Exchange |
|---|---|---|
| CH3NO2 | 10.2 | 120 |
| CH3CH2NO2 | 8.5 | 20 |
| (CH3)2CHNO2 | 7.7 | 1.0 |
Since the nitroalkanes used in this study are stronger acids than water, the kinetic exchange experiments must be conducted under milder conditions than those used for the terminal alkyne. This is achieved by using smaller base concentrations and lowering the temperature of the exchange reaction. Accurate pKa's of 2-nitropropane, nitroethane and nitromethane may be measured directly in aqueous solution. These kinetic and equilibrium acidities are listed in the table on the right. Note that for these three compounds, kinetic acidity changes in an opposite fashion to equilibrium acidity. The kinetic order seems to reflect steric hindrance and carbanion stability; whereas, the equilibria favor increased substitution of the aci-anion double bond.
Base-catalyzed isotope exchange studies of compounds incorporating more
than one set of acidic hydrogens provides additional insight concerning the
creation and use of nucleophilic conjugate bases. Ketones provide many
examples of regioisomeric enolate base formation, and the following diagram
shows two such cases. As noted in the nitroalkane study, hydrogens on an
α-methyl group are exchanged more rapidly than those on more substituted
α-carbon atoms. The equations in the diagram show only the initial product
from a single exchange. These products have additional α-hydrogens which
are also exchanged by subsequent reactions of this kind, so that complete
replacement of all α-hydrogens by deuterium takes place in a short
time.
The relative stability of the resulting enolates increases with
substitution of the enolate double bond. Equations showing the equilibrium
concentrations of these isomeric enolates will be displayed by clicking the
Toggle Equations button. In order to
determine enolate anion equilibria for these ketones, the bulky strong base
sodium hexamethyldisilazide (pKa = 26) was used.
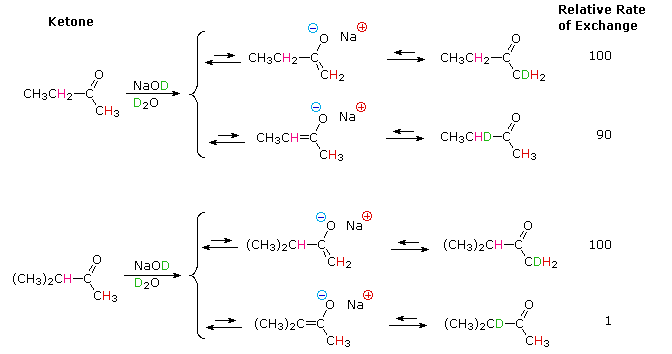
By clicking the Toggle Equations button a second time, the relative rates of α-hydrogen exchange for some substituted cyclohexanones will be displayed above. Once again, less substituted α-carbons exchange more rapidly, but more highly substituted enolates are found to predominate under equilibrium conditions. A third click of the Toggle Equations button will display an energy profile for the 2-methylcyclohexanone case, which should clarify the distinction between kinetic and equilibrium acidity. Two other examples are also shown. These displays may be cycled repeatedly.
Most carbon acids yield conjugate bases that are stabilized by charge delocalization onto neighboring heteroatoms. This resonance stabilization requires significant structural reorganization of the initial compound, which in turn imposes an energy barrier that retards the rate of proton abstraction. For example, the alpha-carbon of a ketone or ester must undergo rehybridization as the enolate anion is formed. The stereoelectronic demands of this change have been described , and it is not surprising that enolate anion formation is much slower than equivalent proton transfers between alcohols and other hydroxylic compounds. Deprotonation rates of phenol and nitromethane, compounds with nearly identical pKa's (10.0), provide an instructive example of this structural reorganization factor. The acidic proton in phenol is bound to oxygen, so deprotonation requires little structure change and is very fast. Nitromethane is a carbon acid. Deprotonation to an aci-anion involves considerable structural change, and is a million times slower than phenolate formation. These structural changes are illustrated in the following diagram.
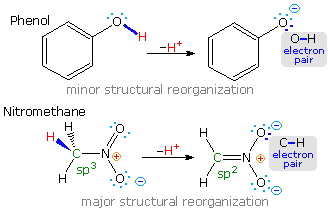
Note that the O-H electron pair in phenol remains largely on oxygen in the corresponding conjugate base, whereas the C-H electron pair in nitromethane is predominantly shifted to oxygen in its conjugate base (colored blue).
The trends outlined here are a bit oversimplified, since solvent and cation influences have been ignored. For a discussion of these factors, and practical applications of enolate anion intermediates in synthesis Click Here.
End of this supplementary topic
Carbon Oxidation States
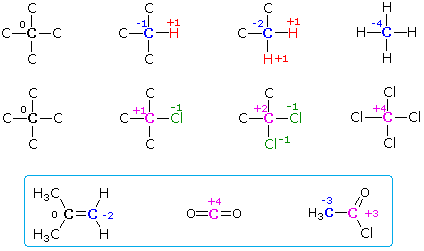
Assigning Oxidation State Numbers to Carbon
The qualitative rules repeated below may be used to create a carbon atom redox number that reflects its oxidation state.
1. If the number of hydrogen atoms bonded to a carbon
increases, and/or if the number of bonds to more electronegative atoms
decreases, the carbon in question has been reduced (i.e. it is in
a lower oxidation state).
2. If the number of hydrogen atoms bonded to a carbon decreases,
and/or if the number of bonds to more electronegative atoms increases,
the carbon in question has been oxidized (i.e. it is in a higher
oxidation state).
3. If there has been no change in the number of such bonds, then
the carbon in question has not changed its oxidation state. In the
hydrolysis reaction of a nitrile shown above, the blue colored carbon has
not changed its oxidation state.
We begin by noting that elemental carbon has a zero oxidation state by definition. A carbon atom bonded only to other carbon atoms, as in the structures on the right below, is therefore assigned an oxidation number of zero. If one of the carbon substituents is replaced by a hydrogen atom this oxidation number changes by -1; whereas, if the carbon substituent is replaced by a more electronegative substituent (O, N, F, Cl, Br etc.) the oxidation number changes by +1. Further examples of this simple system are shown in the diagram.
End of this supplementary topic
Stereoelectronic Effects
Stereoelectronic Effects
Factors that influence the properties or reactivity of molecular species
as a consequence of the spatial orientation of filled or unfilled orbitals
are termed stereoelectronic. In many cases, electron pairs in atomic
or molecular orbitals that are involved in the making or breaking of bonds
have an optimal geometrical alignment that is critical for a reaction to
occur. This alignment provides the best overall bonding of participating
species during the course of a reaction, and reflects the fact that
transition states having the greatest bonding energy have lower potential
energies than transition states with less bonding.
Two relatively simple examples of stereoelectronic effects are found in
SN2 and E2 reactions. The mechanisms of these reactions were
described earlier in our discussion of alkyl halide
chemistry, but a more extensive examination of these common
transformations will provide a helpful introduction to this
subject.
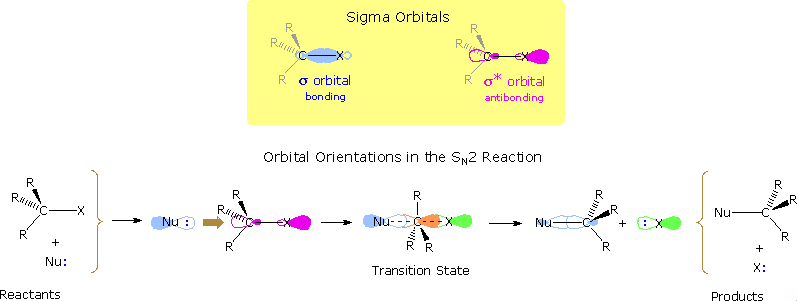
1. SN2 Reactions
Nucleophilic substitution reactions of alkyl halides take place by a continuum of mechanisms, defined by SN2 behavior at one extreme and SN1 behavior at the other. The essential characteristics of the SN2 process include inversion of configuration at the reaction site, sensitivity to steric hindrance in the alkyl group, and second order kinetic behavior. The orbital interactions that take place in a successful SN2 reaction are shown in the following diagram. The bonding and antibonding sigma molecular orbitals composing the C-X bond are drawn in the yellow box at the top of the diagram. As a nucleophile approaches the rear side of the carbon, the orbital containing the non-bonded electron pair (light blue) begins to overlap with the empty antibonding C-X sigma orbital (pink colored), shown on the left of the bottom line. As electrons occupy this antibonding orbital, the C-X bond is weakened. In the transition state partial bonds exists between the carbon and both the nucleophile and the halogen (the carbon is essentially sp2 hybridized with the orange colored p-orbital providing the partial bonding). Finally, a full C-Nu sigma bond develops and the halogen leaves as an anion.
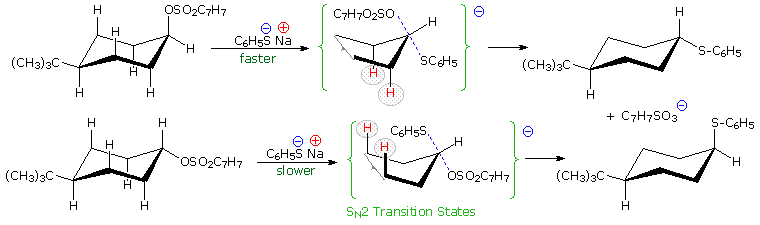
Other substitution reactions also proceed by the SN2 pathway, as illustrated by the stereoisomeric cyclohexyl tosylates in the following diagram. The bulky tert-butyl group on C-4 serves to lock the chair conformation in the configuration shown, so one isomer has an equatorial tosylate function, and the other an axial tosylate. On reaction with sodium thiophenolate, both isomers undergo bimolecular substitution with inversion, the axial isomer reacting about thirty times faster than its equatorial epimer. Steric hindrance to rear side nucleophile approach by the red colored hydrogen atoms is present for each isomer, but the relief of steric crowding of the axial tosylate leaving group helps to facilitate substitution of the that isomer.
SN2 reactions may be intermolecular, as in these examples, or
intramolecular. By clicking on the above
equations, an example of two sequential substitutions, the first
intermolecular and the second intramolecular, will be displayed. Note that
the intramolecular substitution shown here proceeds via the same orbital
alignment described above. The reacting moieties in intramolecular
reactions are incorporated in the same molecule; consequently, such
reactions exhibit first order kinetic behavior instead of the usual second
order kinetics. Because enolate
alkylation reactions are irreversible, the strained four-membered ring
product is stable under the reaction conditions.
In general, intramolecular forms of bimolecular reactions are faster
than their intermolecular counterparts, since the reactive sites are
held close together (their relative concentrations are as high as
possible). The rapid lactonization
of gamma and delta-hydroxy acids compared with corresponding
intermolecular esterifications is another example of this principle, which
is associated with a favorable entropy change. Note,
however, that ring size has a profound influence on
the intra- vs. intermolecular dichotomy.
Maximum overlap of the electron orbital of the nucleophile with the
antibonding sigma orbital of the carbon substituent requires an approach
from the rear side of the carbon, ideally 180º from the leaving group. The
importance of this orientation, which is shown in previous diagrams, was
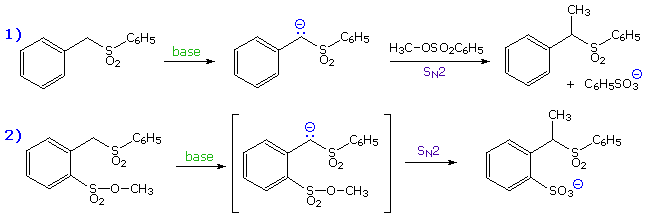
made clear by an experiment conducted by A. Eschenmoser over 25 years ago. The
results of Eschenmoser's experiment are presented in the following
illustration. Initially, two equations are written. The first describes the
intermolecular methylation of a sulfone anion by methyl tosylate, a typical
SN2 reaction. The second is a very similar reaction which many
would consider to be intramolecular, since a methyl sulfonate moiety is
positioned ortho to the sulfone function.
By clicking on these equations, evidence against
the intramolecular mechanism will be displayed. Although intramolecular
reactions are often favored, this case requires a significant departure
from the 180º orbital alignment required by an authentic SN2
reaction. Since this reaction was found to be second order and is not
faster than the simple intermolecular analog (reaction 1 on the
preceding slide), it cannot be intramolecular.
2. E2 Reactions
Elimination of vicinal groups, usually called 1,2- or beta-elimination, is the most common type of elimination reaction. Examples of some typical cyclohexyl halide eliminations are given in the following illustrations. From the location of the double bond in the elimination products, and the relative rates of elimination, it is clear that a diaxial orientation of the eliminating atoms or groups (colored red) is necessary for optimum reaction.
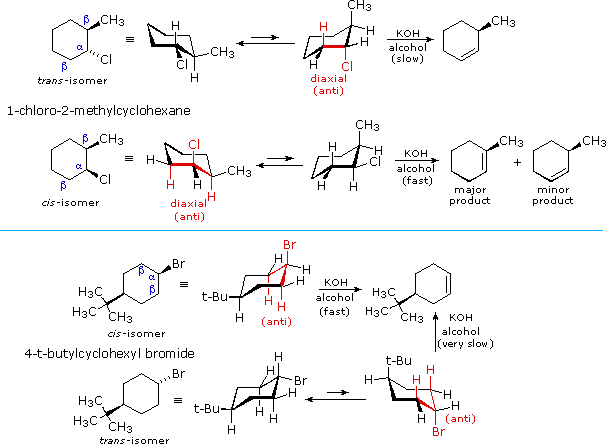
|
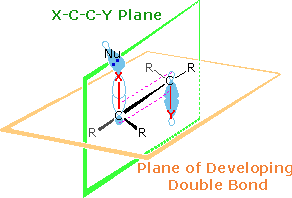
Two views of the characteristic orbital orientations of this
anti-elimination are drawn on the right. Two planes are defined in
the upper drawing. The light green plane defines the coplanar nature of the
leaving groups (X & Y) and the carbons to which they are bonded. The
light orange plane identifies the plane of the final double bond. Together
with the lower drawing, the initial display shows the overlap of
participating bonding orbitals. The corresponding antibonding orbitals of
the C-X and C-Y bonds will appear by clicking on the
upper display. This orbital alignment defines the stereoelectronic
character of these elimination reactions. Note that the "anti" descriptor
for this configuration refers to the antarafacial
relationship of X and Y with respect to the light orange plane. Also, the
E2 designation refers to the second order kinetics observed for these
reactions.
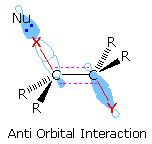
Although anti-eliminations are favored when possible, syn-elimination is
observed in some cases. By clicking on the lower
display, it will be replaced by a similar orbital drawing for syn
elimination. In both syn and anti-transition states the X-C-C-Y bonds are
coplanar, an alignment that allows facile conversion of two sigma bonds
(colored red) to a pi-bond. Because of the eclipsed configuration of the
syn-transition state, it is less stable than a corresponding
anti-transition state. Syn-elimination is sometimes observed when Y is an
ionic species, such as (CH3)3N(+); but it
usually occurs when an anti configuration is unstable or not possible, as
in the following example. Isotopic substitution confirms that this
base-catalyzed beta-elimination takes place by a syn mechanism.
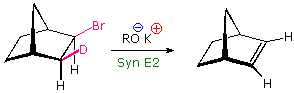
The anti-configurational relationship of the leaving groups in E2 reactions is relatively easy to see in the cyclohexyl compounds shown above, thanks to the chair conformations of these substrates. If this stereoelectronic feature is general for all E2 reactions, it should also hold for eliminations of acyclic compounds. The following examples illustrate this behavior. Conformational mobility about the central C-C bond allows each stereoisomer to adopt an anti alignment of leaving groups (colored green), and this is reflected in the products.
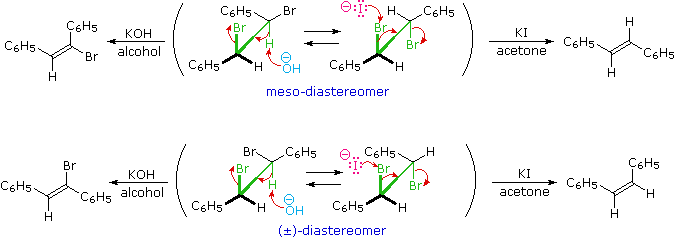
3. Trans-Addition to Alkenes
Addition reactions to carbon-carbon double bonds show a parallel stereoselectivity to the elimination reactions. Both syn and anti additions are observed, as noted in an earlier chapter. Indeed, the trans-addition of halogen reagents provides good examples of the importance of stereoelectronic control, even in the presence of opposing steric factors. The addition of bromine to 4-tert-butylcyclohexene, shown here, is a simple case. Assuming trans-addition, two stereoisomeric products are possible. Remarkably, the product having two axial bromines is favored over its diequatorial isomer. In order to maintain maximum bonding throughout the addition, carbon-bromine bonds must be aligned perpendicular to the plane of the double bond. This diaxial orientation mirrors the stereoelectronic demands of the E2 eliminations discussed above, and may be realized in two ways.
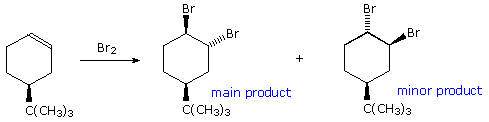
By clicking on the equation, the first and
most favored addition will appear above. A half-chair conformation for the
substituted cyclohexene is drawn on the left. The large t-butyl substituent
occupies an equatorial orientation and serves to hold the conformer as
shown. Trans-addition in the manner depicted leads directly to the diaxial
product. Clicking on the drawing a second time
displays the alternate trans addition. In order to maintain good orbital
alignment throughout this reaction, the cyclohexane ring must adopt a
higher energy twist-boat conformation. This on relaxing to a chair gives
the diequatorial product. As a result, the first addition has a lower
activation energy than the second.
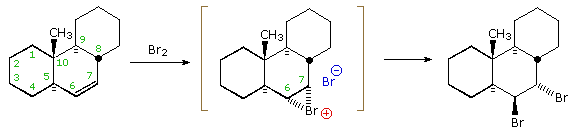
The attentive reader will recall that trans-addition of bromine and other
halogen reagents was attributed to nucleophilic ring opening of a cyclic halonium
ion intermediate. It is instructive to consider this aspect of the
stereoelectronic factor using a somewhat more rigid alkene substrate. This
is shown in the following illustration. The trans-fused six-membered rings
hold the central cyclohexene ring in a tight configurational grip.
Chair-chair interconversions are not possible, and even twist-boat
conformers are very strained. The initial electrophilic attack of bromine
takes place from the underside of the molecule, due to the steric hindrance
of the axial methyl group. The resulting bromonium ion is written in
brackets, and it is apparent that nucleophilic attack from the top would be
less hindered at C-7 than at C-6 (the methyl group again). By clicking on the equation, a conformational drawing of this
reaction will be displayed. The two possible sites of attack are designated
by green (C-6) and red (C-7) arrows. Although bromide ion attack at C-6 is
more hindered, it is favored by the diaxial stereoelectronic factor, and
this is the initially observed product. Subsequent rearrangement of this
diaxial dibromide to its diequatorial isomer takes place slowly.
Stereoelectronic control of epoxide ring opening follows the same principles.
4. Addition of Nucleophiles to Carbonyl Groups
Stereoelectronic factors have been shown to influence the addition of nucleophilic reagents to carbonyl groups, particularly for aldehydes and ketones. An outline of this stereoelectronic effect is provided in the following diagram. The essential molecular orbitals are drawn to the left of the diagram, and the initial bonding with a nucleophile is believed to take place with the empty antibonding pi-orbital. This would explain the favored bonding alignment, known as the Bürgi-Dunitz trajectory.
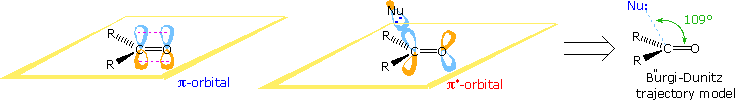
Many carbonyl addition reactions have been analyzed by a combination of steric and stereoelectronic factors. In the following illustration, two hydride reductions of methyl ketones are shown. Since each ketone has an existing stereogenic site, and since the reduction creates a new chiral center, diastereomeric products are possible. These may be designated in several ways, but the syn-anti notation is generally preferred. When the existing stereogenic center is located next to the carbonyl group, as in the upper equation, it may influence the proportion of product diastereomers. Diastereoselectivity of this kind is sometimes called asymmetric induction. This influence is, of course, the same for both an enantiomerically pure or a racemic reactant. If, however, the stereogenic center is far away from the carbonyl group, it has a negligible influence on the reduction, and a 50:50 mixture of diastereomers is produced (lower example).
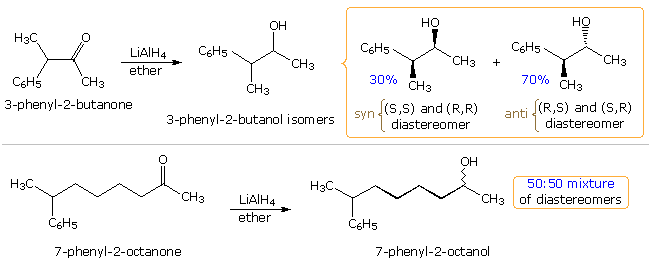
A number of models have been proposed to explain the
diastereoselectivity of the first reduction. Many conformations about the
alpha-C-CO bond may be written, and the challenge is to pick one that
accounts for the observed selectivity. By clicking on
the drawing, three of these will be displayed. For general use, the
substituents on the chiral center adjacent to the carbonyl group are
labeled L (large), M (medium) and S (small) to reflect
their size. In each model the sterically preferred Bürgi-Dunitz approach is
shown by a pink arrow, and each predicts the correct configuration of the
favored diastereomer. The earliest rationalization was offered by D. Cram and is shown by the model on the
left. A more favored conformation was chosen by G. Karabatsos, as shown by
the center model. The most recent model (on the right) is that proposed by
H. Felkin. These models all require classification of S, M & L
substituents, occasionally a tricky process, and assume a reactant-like
transition state for the reduction. Even so, some bonding of hydride to the
carbonyl carbon must take place in the transition state, accompanied by
corresponding small structural changes. Plausible structures for these
transition states are given in the orange box. An important point needs to
be made here.
The ratio of products obtained from a a group of
equilibrating conformers is determined by transition state energies, not
conformer concentrations.
This generally useful rule is called The
Curtin Hammett Principle.
A brief analysis of these models is instructive. The Cram model makes use of an unfavorable conformer (R & L are eclipsed). Although this strain is slightly relieved in the transition state, the oxygen and associated metals is moving toward the M group, resulting in increased crowding. The Karabatsos model starts with a favorable conformer, but the nucleophile trajectory nearly eclipses the C-S bond (~20º dihedral angle). The oxygen shift in the transition state relieves eclipsing strain with M, benefiting the transition state energy. Finally, the Felkin-Ahn model seems to offer the best rationalization. The nucleophile trajectory is roughly 40º away from eclipsing the C-S bond, and both the oxygen and the R group undergo favorable shifts in the transition state.
Cyclic ketones have fewer low energy conformations than do acyclic ketones, and diastereoselectivity in nucleophilic addition reactions is often observed. An analysis similar to that used above for the acyclic compounds has proven useful in explaining and predicting the outcome of such reactions. By clicking on the drawing a second time, the hydride reduction of 2-methylcyclopentanone will be displayed. The trans isomer is favored by a 3:1 ratio. Again, steric hindrance to hydride approach on the Bürgi-Dunitz trajectory rationalizes this finding.
|
For two additional examples of stereoelectronic effects in conformational equilibria Click Here. |
|---|
5. Stereoelectronic Modulation of Cyclization
The formation of rings from suitably substituted molecular chains is a common requirement in synthesis. In general terms this transformation takes place by the bonding together of two reactive functional groups located at the ends of a chain. In the following diagram these are represented by an electrophilic group (E), and a nucleophilic group (Nu). Two possible modes of reaction are outlined here. In order to form a ring, an intramolecular reaction must take place. However, any significant quantity of reactant contains approximately 1020 molecules, and these may react with each other in an intermolecular manner to give dimeric and larger products. Controlling the course of such a reacting system is not a trivial matter, and many factors influence the outcome. Among these, the thermodynamic functions enthalpy and entropy are especially important.
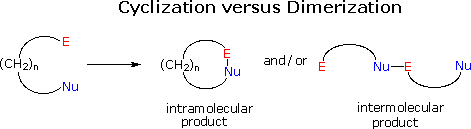
The enthalpy factor is associated with ring strain, which
includes angle and eclipsing strain. Small three and four-membered rings
are destabilized
thermodynamically by such strain, and as a rule cannot be prepared by
reversible bond formation. Five and six-membered rings are less strained,
and are often created by reversible bond formation, as in the cases of
lactonization
of hydroxy acids and Dieckmann
condensations. Medium sized rings (8 to 11 atoms in size) are generally
destabilized by transannular
crowding (steric hindrance by groups on opposite sides of the
ring).
The entropy factor is related to the concentration of chain conformations
in which the ends to be joined are close together. For a three carbon chain
this concentration is nearly 100%, but it drops off rapidly as the length
of the chain increases. The number of different conformers a chain may
assume increases with the length of the chain, and the proportion of
conformers having proximate ends decreases. These thermodynamic factors
interact in opposition to each other. Entropy favors small ring formation,
even though this is energetically (enthalpically) unfavorable. Irreversible
bond forming reactions, such as enolate alkylations, are used to achieve this outcome. Five and six-membered ring formation is
favored by both factors, and may be achieved by both reversible and
irreversible bond formation. The often cited principle that
intramolecular reactions are favored over intermolecular reactions
has its origin here. Medium and large sized rings, on the other hand, are
difficult to make due to the low probability of suitable conformers in the
reaction mixture. Intermolecular dimerization and polymerization are
usually observed unless the reactions are conducted at high dilution.
Remember, the rate of a
bimolecular reaction, such as dimerization is proportional to the
square of reactant concentration (2nd order); whereas, the unimolecular
cyclization reaction has a rate proportional to the first power of reactant
concentration (1st order). Thus, at very low reactant concentration,
cyclization occurs faster than dimerization.
Because of the structural constraints in a cyclization transition state, stereoelectronic influences may be anticipated. One example is shown by the following two equations. Conjugate addition of amines to unsaturated carbonyl reactants is usually rapid and reversible, as demonstrated by the first equation. At first glance, the reactant in the second equation might be expected to undergo an intramolecular reaction of the same kind, yielding a five-membered heterocyclic ester (drawn in the red box). In practice, however, the reaction is slower and takes a different course, producing a five-membered lactam by direct acylation of the amine by the ester.
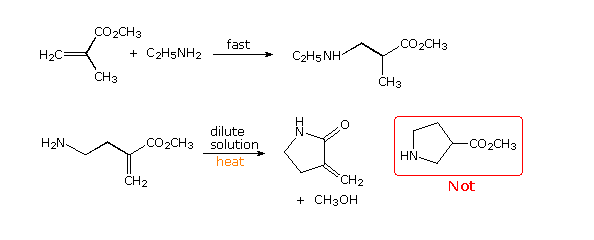
To explain this unexpected change in behavior, we must examine the orbital trajectories permitted in the two transition states (1,4- versus 1,2-addition). By clicking on the drawing a representation of these transition states will be displayed. Neither achieves a perfect Bürgi-Dunitz approach of the nitrogen nucleophile to a double bond, but the 1,2-addition comes much closer. Furthermore, the best 1,4-addition trajectory is unable to occur in a plane orthogonal to the plane of the double bond; whereas, the 1,2 addition is again much closer to the ideal orientation. For a display of 1,4- versus 1,2-addition in this example Click Here.
6. The Baldwin Rules
General stereoelectronic constraints on cyclization reactions were
recognized by J. Baldwin, and summarized in a qualitative model now
referred to as the Baldwin Rules. The following diagram illustrates
the five major cyclization modes treated by Baldwin. In each case a
nucleophilic moiety bonds to an electrophilic site (usually a carbon atom),
designated by a purple dot. The flow of electrons during bond formation may
be encompassed by the developing ring (an endo cyclization), or may shift
away from the ring (an exo cyclization). Furthermore, the electrophilic
site may have different hybridization states. The terminology used to
identify a given ring formation consists of three parts: the ring size,
given by n + 3 in the diagram (where n=0 or an integer), the exo / endo
feature, and the hybridization. Since it is unlikely that endo-tet
reactions will be useful for making rings, such reactions are not included
here. However, the possibility of a six-membered cyclic analog of this kind
was discussed earlier.
Using this terminology, the previous cyclization of the amino ester to a
lactam is a 5-exo-trig process (the double bond is the carbonyl
group). The alternative ring closure by conjugate addition is
5-endo-trig, with respect to the carbon-carbon double bond,
even though the conjugative shift of electrons to the ester carbonyl is
exocyclic.
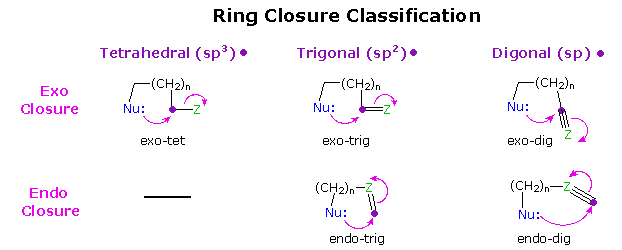
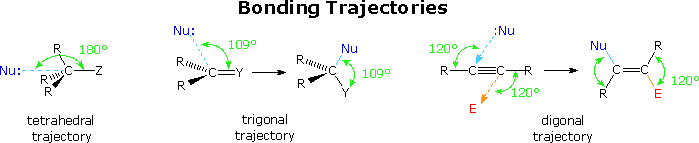
The stereoelectronically favored trajectories for nucleophilic bonding to carbon atoms of different hybridization states are summarized below. The tetrahedral and trigonal cases have already been described. It is clear that these trajectories represent a "least motion" transition state in which bonding is kept to a maximum throughout the reaction. By applying these trajectories to the five cyclization categories noted above, Baldwin was able to discern the effect of ring size on an idealized transition state. In some cases the constraints of a given ring size perturbed the transition state to such a degree that reaction seemed improbable. Such cases were called unfavorable.
The following table summarizes Baldwin's conclusions. The 3-endo-trig,
4-endo-trig and 5-endo-trig combinations, together with the 3-exo-dig and
4-exo-dig possibilities (all colored pink in the table) were classified as
"unfavorable". All the other combinations were classified "favorable", and
are colored light green in the table.
Remember, this evaluation is based primarily on stereoelectronic factors.
So called favorable cyclizations may fail for other reasons, and sometimes
an unfavorable cyclization may take place when alternative reactions are
even less favored.
Ring Closure Categories
| Tetrahedral Closure | 3-exo-tet | 4-exo-tet | 5-exo-tet | 6-exo-tet | 7-exo-tet |
|---|---|---|---|---|---|
| Trigonal Closure | 3-exo-trig | 4-exo-trig | 5-exo-trig | 6-exo-trig | 7-exo-trig |
| 3-endo-trig | 4-endo-trig | 5-endo-trig | 6-endo-trig | 7-endo-trig | |
| Digonal Closure | 3-exo-dig | 4-exo-dig | 5-exo-dig | 6-exo-dig | 7-exo-dig |
| 3-endo-dig | 4-endo-dig | 5-endo-dig | 6-endo-dig | 7-endo-dig | |
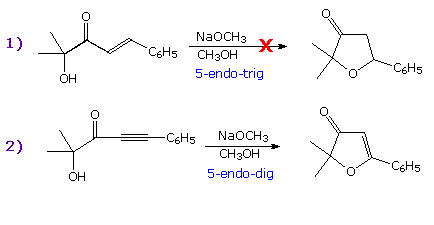
In the examples on the right the first equation describes a 5-endo-trig
cyclization that fails to take place. The second equation shows a
successful 5-endo-dig reaction, similar in many respects to the first.
When an enolate anion serves as the nucleophilic agent in a ring
closure, a modification of the Baldwin rules should be used. These rules
will be shown on the right by clicking on the
drawing. An example comparing a 5-(enolexo)-exo-tet closure with an
isomeric 5-(enolendo)-exo-tet option will then replace the modified rules
by a second click on the drawing. A third click on the drawing will display a similar
6-(enolendo)-exo-trig example.
7. Rationalizing Conformational Preferences
The following diagram shows two conformational equilibria in which one
of the isomers is favored. Esters generally prefer to adopt a s-Z
conformation (sometimes termed s-trans), as shown in the first example.
This notation, which refers to the alkoxy sigma-bond to the carbonyl
carbon, corresponds to that used for diene
conformations. Lactones incorporated in seven membered and smaller
rings are forced into an s-E conformation, and consequently are more
reactive.
In the second example, tri-tert-butyl hexahydro-(1,3,6)-triazine exists
predominantly in a configuration having one axial tert-butyl group.
Explaining these structural preferences provides an instructive review of
stereoelectronic factors.
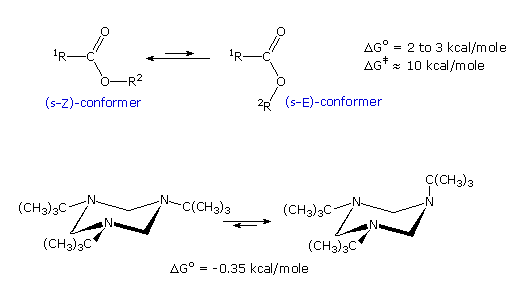
The ester equilibrium is most simply analyzed by considering electron
and dipole interactions. The latter will be displayed by clicking on the diagram. In the s-E conformer the carbonyl
group dipole is repelled by the similarly oriented ether oxygen dipole,
whereas these dipoles have opposite directions in the s-Z conformer. Also,
the non-bonded electron pairs of the two oxygens are closer together in the
s-E conformer than in the s-Z conformer. A similar analysis of the second
example shows a parallel alignment of all three amine dipoles in the all
equatorial conformer, and a less repulsive arrangement in the axial
conformer.
A thorough evaluation of these cases should also examine the interaction of
a non-bonding electron pair with neighboring (vicinal) antibonding orbitals
that may function as electron acceptors. This analysis of the ester
equilibrium will be shown by clicking on the
diagram a second time. The upper part of the diagram illustrates the
p-pi conjugation of one non-bonding electron pair with the carbonyl group.
This conjugation explains the large energy barrier for interconversion of
the E & Z conformers. The remaining electron pair occupies a
sp2 orbital (colored pink), and this is the donor pair that may
have a bonding interaction with an acceptor orbital (an anomeric
effect). Only the Z-conformer provides this stabilizing interaction,
which involves the antibonding C-O orbital (light gray) of the carbonyl
sigma bond (colored red). A similar donor-acceptor interaction provides
additional stabilization for the axial-tert-butyl conformer in the second
example, as illustrated by clicking on the
diagram a third time.
8. The Anomeric Effect
The anomeric effect was first recognized in carbohydrate chemistry, but applies generally to all organic compounds.
End of this supplementary topic
Carbocation Stability
Factors Influencing Carbocation Stability
Carbocation intermediates, sometimes called carbonium ions, have been proposed or identified as important species in reactions ranging from electrophilic addition to alkenes, to unimolecular solvolysis of alkyl halides. The relative stability of these cationic intermediates varies markedly with the presence of substituents on the trivalent charged carbon atom, in a fashion that has led useful empirical rules for predicting reaction selectivity (summarized elsewhere). This stability order for simple alkyl alkenyl and aryl substituted carbocations is repeated below, followed by a table of supportive data.
|
|
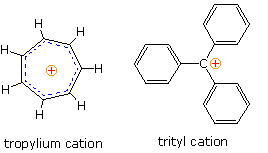 A few carbocations, such
as tropylium and trityl (triphenylcarbenium) shown on the right, are
sufficiently stable to form isolable salts with poorly nucleophilic anions,
such as tetrafluoroborate (BF4(–) ). However, most
carbocations are unstable and very reactive under normal laboratory
conditions, so conventional studies of all but the most stable of these
species have not been possible. Nevertheless, gas phase ionization energies
of alkyl chlorides, hydride affinity measurements (gas phase), molecular
orbital calculations, and low temperature nmr
examination of ionized alkyl halides in mixed solvents composed of
SbF5, SO2, SO2F2 &
SO2FCl. (referred to as "super acids") have confirmed the
qualitative relationship shown above. At low temperatures, 1H
and 13C nmr spectra of (CH3)3C(+) and
(CH3)2CH(+) were obtained and interpreted. The
charged tricoordinate carbon atom exhibited a 13C signal over
300ppm downfield from TMS.
A few carbocations, such
as tropylium and trityl (triphenylcarbenium) shown on the right, are
sufficiently stable to form isolable salts with poorly nucleophilic anions,
such as tetrafluoroborate (BF4(–) ). However, most
carbocations are unstable and very reactive under normal laboratory
conditions, so conventional studies of all but the most stable of these
species have not been possible. Nevertheless, gas phase ionization energies
of alkyl chlorides, hydride affinity measurements (gas phase), molecular
orbital calculations, and low temperature nmr
examination of ionized alkyl halides in mixed solvents composed of
SbF5, SO2, SO2F2 &
SO2FCl. (referred to as "super acids") have confirmed the
qualitative relationship shown above. At low temperatures, 1H
and 13C nmr spectra of (CH3)3C(+) and
(CH3)2CH(+) were obtained and interpreted. The
charged tricoordinate carbon atom exhibited a 13C signal over
300ppm downfield from TMS.
1. Inductive & Resonance Effects
What are the factors that influence carbocation stability? The most common means of stabilizing an ion is by charge delocalization, either by inherent structural interactions or by solvation. As noted elsewhere, such structural interactions may usually be classified as inductive or resonance effects, and these may complement or oppose each other. Examples of both are given in the following diagram.
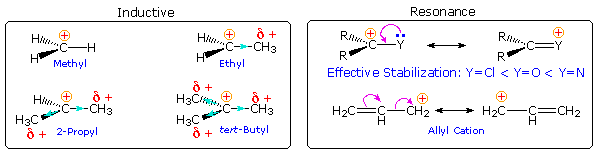
Alkyl groups have somewhat lower electronegativities and are more
polarizable than hydrogen. If an alkyl group is bonded to the carbocation
center, the electron pair of the C-C sigma bond will shift toward the
positive charge, transferring a small part of that charge to the alkyl
group. In the diagram on the left above, this inductive electron shift is
designated by a light blue arrow head. Additional alkyl groups provide
increased inductive charge dispersion, with each group assuming a share of
the charge. Clearly, this analysis supports the stabilizing influence of
alkyl substituents on carbocations.
Resonance stabilization by non-bonding electron pairs on adjacent
heteroatoms is particularly strong, as shown on the right above. Such
charge delocalization overcomes the potential inductive destabilization of
these electronegative substituents. Similar stabilization is provided by an
adjacent nucleophilic pi-electron function, such as a double bond. A phenyl
substituent affords even greater charge delocalization than a double bond,
as reflected by the position of a benzyl cation in the stability order.
Resonance stabilization is generally stronger than inductive effects, and
is the predominant factor stabilizing the tropylium and trityl
cations.
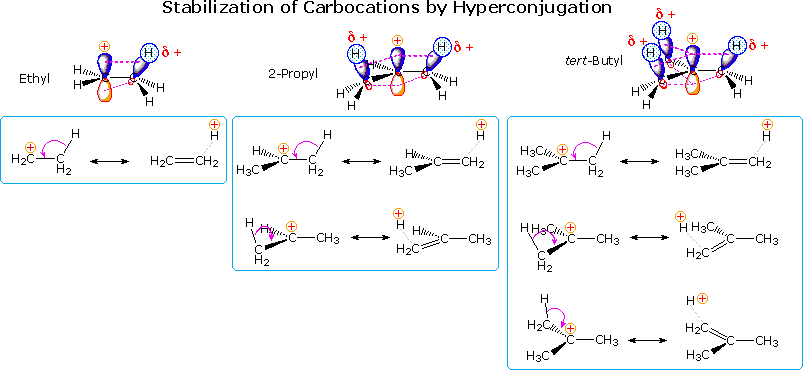
2. Hyperconjugation
Another way in which alkyl substituents stabilize carbocations is illustrated in the following diagram. This conjugative charge delocalization, called hyperconjugation, involves partial pi-bond formation to alpha-carbon atoms, provided suitably oriented C-H or C-C bonds are present. The small increase in stability of the 1-propyl cation compared with an ethyl cation, as noted above, suggests that C-C hyperconjugation provides slightly greater stabilization than does the C-H hyperconjugation shown here. Hyperconjugation by alkyl substituents also acts to stabilize unsaturated functional groups, as noted earlier for carbon-carbon double bonds.
Since hyperconjugation and the inductive effect act in the same manner,
their relative importance in carbocation stabilization is a matter of
interest. Some insight to this question is found in a group of novel
compounds, bridgehead substituted bicyclic halides. Two examples of these
compounds are shown below. The nomenclature of bridged bicyclic compounds
identifies the length of the chains that connect the bridgehead atoms
(colored pink here). Three connecting chains are present in a bicyclic
compound, and the number of atoms in each chain (excluding hydrogen) is
given as a number in brackets. If the chains are of different lengths the
longest is listed first. One of the bridgehead atoms is numbered one, and
the longest chain continues the numbering sequence until the second
bridgehead atom is included. Numbering then continues along the next
longest chain. The base name of the bicyclic compound reflects the total
number of carbons, and is therefore the sum of the bridging chains plus two
(the bridgehead atoms).
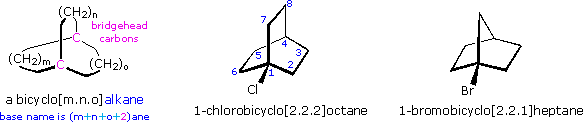
The bridgehead substituted halides shown above will form 3º-carbocations
when ionized. Inductive stabilization of these cations should be similar to
that of the tert-butyl cation, so if this were the predominant stabilizing
factor from alkyl substitution, the reactivity of these halides should be
similar to their tert-butyl counterparts. In practice, however, the
bridgehead halides were found to be much less reactive. Indeed,
1-chlorobicyclo[2.2.1]heptane was recovered unchanged from prolonged
treatment with hot ethanolic
silver nitrate. The instability of such bridgehead carbocations has
been attributed to the pyramidal shape forced upon the trigonal carbon
(sp2 hybridized). However, covalent bonds are generally able to
accommodate modest bending distortions without significant destabilization,
and the inductive shift of electron pairs toward the positively charged
carbon atom is unlikely to be impeded by a pyramidal configuration of the
carbocation.
An alternative explanation is that hyperconjugation with the
alpha-methylene groups is prohibited by the rigid configuration of these
bridgehead cations. By clicking on the diagram
above, an illustration of one possible C-H hyperconjugative
interaction will be displayed. The carbon-carbon double bond implicit to
this occurrence is badly twisted, and could not exist as such in any stable
alkene. Structural prohibitions of this kind are encompassed in an
empirical guideline called Bredt's
rule.
On the other hand, C-C hyperconjugation may act to partially stabilize
bridgehead carbocations. By clicking on the
diagram a second time, two examples of such hyperconjugation will
appear. While still relatively inert, the bicyclo[2.2.2]octane compound is
roughly a million times more reactive than its bicyclo[2.2.1]heptane
analog, shown above. This may be attributed to improved hyperconjugation,
since the appropriate C-C bonds (colored red) are better aligned with a
developing bridgehead carbocation. Furthermore, confirmation of expected
changes in bond lengths resulting from such hyperconjugation has been
obtained by X-ray diffraction analysis of a crystalline SbF6
salt of the adamantane cation. In this study, the red-colored bonds were
lengthened and the green-colored bonds were shortened.