An Introduction to Synthesis
The study of organic chemistry exposes a student to a wide range of
interrelated reactions. Alkenes, for example, may be converted to
structurally similar alkanes, alcohols, alkyl halides, epoxides, glycols
and boranes; cleaved to smaller aldehydes, ketones and carboxylic acids;
and enlarged by carbocation and radical additions as well as
cycloadditions. All of these products may be transformed subsequently to a
host of new compounds incorporating a wide variety of functional groups,
and thereby open to even further elaboration. Consequently, the logical
conception of a multistep synthesis for the construction of a designated
compound from a specified starting material becomes one of the most
challenging problems that may be posed.
A one or two step sequence of simple reactions is not that difficult to
deduce. If, for example, one is asked to prepare meso-3,4-hexanediol from
3-hexyne, most students realize it will be necessary to reduce the alkyne
to cis or trans-3-hexene before undertaking glycol formation. Permanaganate
or osmium tetroxide hydroxylation of cis-3-hexene would form the desired
meso isomer. From trans-3-hexene it would be necessary to first epoxidize
the alkene with a peracid, followed by ring opening with hydroxide ion.
This example illustrates a common feature in synthesis: often there is
more than one effective procedure that leads to the desired
product.
Longer multistep syntheses require careful analysis and thought, since
many options need to be considered. Like an expert chess player evaluating
the long range pros and cons of potential moves, the chemist must appraise
the potential success of various possible reaction paths, focussing on the
scope and limitations constraining each of the individual reactions being
employed. This can be a daunting task, the skill for which is acquired by
experience, and often trial and error.
The three examples shown below are illustrative. The first is a simple
functional group conversion problem, that may initially seem difficult. It
is often helpful to work such problems backwards, starting from the
product. In this case it should be apparent that cyclohexanol may be
substituted for cyclohexanone, since the latter could then be made by a
simple oxidation. Also, since cyclohexane (and alkanes in general) is
relatively unreactive, bromination (or chlorination) would seem to be an
obvious first step. At this point one is tempted to convert
bromocyclohexane to cyclohexanol by an SN2 reaction with
hydroxide ion. This reaction would undoubtedly be accompanied by E2
elimination, so it would be cleaner, although one step longer, to first
make cyclohexene and then hydrate it by any of several methods (e.g.
oxymercuration and hydroboration) including the one shown by clicking on the diagram
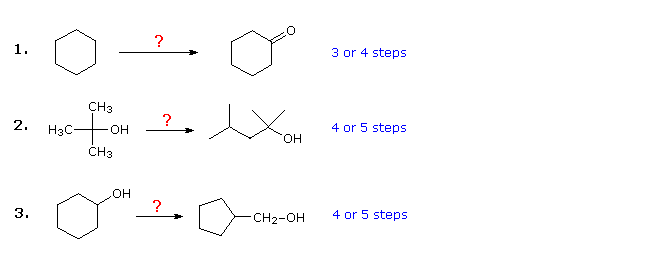
Plausible solutions for the second and third problem will also appear above at this point. In problem 2 the desired product has seven carbon atoms and the starting material has four. Clearly, two intermediates derived from the starting compound must be joined together, and one carbon must be lost, either before or after this bonding takes place. The 3º-alcohol function in the product suggests formation by a Grignard addition to a ketone, and isobutene appears to be a good precursor to each of these reactants, as shown. The reactant and product compounds in the third problem are isomers, but some kind of bond-breaking and bond-making sequence is clearly necessary for this structural change to occur. One possible procedure is shown above. Acid-catalyzed rearrangement of cyclohexene oxide, followed by reduction might also serve.
The useful approach of working out syntheses starting from the target molecule and working backward toward simpler starting materials has been formalized by Prof. E. J. Corey (Harvard) and termed retrosynthetic analysis. In this procedure the target molecule is transformed progressively into simpler structures by disconnecting selected carbon-carbon bonds. These disconnections rest on transforms, which are the reverse of plausible synthetic constructions. Each simpler structure, so generated, becomes the starting point for further disconnections, leading to a branched set of interrelated intermediates. A retrosynthetic transform is depicted by the => symbol, as shown below for previous examples 2 & 3. Once a complete analysis has been conducted, the desired synthesis may be carried out by application of the reactions underlying the transforms.
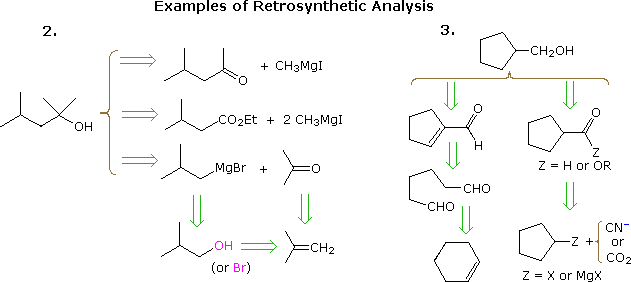
The above diagram does not provide a complete set of transforms for
these target compounds. When a starting material is specified, as in the
above problems, the proposed pathways must reflect that constraint. Thus
the 4-methyl-2-pentanone and 3-methylbutyrate ester options in example 2,
while entirely reasonable, do not fit well with a tert-butanol
start. Likewise, a cyclopentyl intermediate might provide an excellent
route to the product in example 3, but does not meet the specified
conditions of the problem.
Retrosynthetic analysis is especially useful when considering relatively
complex molecules without starting material constraints. If it is conducted
without bias, unusual and intriguing possibilities sometimes appear.
Unfortunately, molecular complexity (composed of size, functionality,
heteroatom incorporation, cyclic connectivity and stereoisomerism)
generally leads to very large and extensively branched transform trees.
Computer assisted analysis has proven helpful, but in the end the instincts
and experience of the chemist play a critical role in arriving at a
successful synthetic plan. Some relatively simple examples, most having
starting material restrictions, are provided below.
Problem 1
A synthesis of N-ethyl-2-aminomethylspiro[3.3]heptane from starting compounds having no more than three contiguous carbon atoms is required. This provides a good example of the importance of symmetry in planning a synthesis. First, it should be recognized that the amine group is best introduced at the end of the synthesis, by reacting ethylamine with an ester (or acyl chloride derivative) of spiro[3.3]heptane-2-carboxylic acid, followed by LiAlH4 reduction. This approach avoids the necessity of protecting a nucleophilic nitrogen from undesired participation in other reactions. Second, the symmetry of the remaining carbon skeleton suggests its disconnection into 1,3-difunctionalized propane units, as shown below. All of these have a common origin in diethyl malonate, which can be reduced to a 1,3-glycol and then converted into 1,3-dibromopropane.
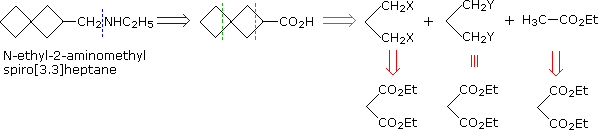
Problem 2
A synthesis of 2,7-dimethyl-4-octanone from starting compounds having no
more than four contiguous carbon atoms is required. The structural formula
and a first-stage retroanalysis of this ketone are displayed in the
following diagram. Three straightforward disconnections are shown, as drawn
by the dashed lines. The first (magenta arrow) is undoubtedly the simplest,
since a Grignard reagent addition to a suitable nitrile gives the product
directly. However, one or more of the reactants is larger than
C4 and must therefore be prepared independently before use. A
two-step procedure involving Grignard addition to an aldehyde, followed by
oxidation of the 2º-alcohol product, also suffers the same requirement, as
do the epoxide opening routes presented in the second row (cyan arrow).
Secondary preparations of these intermediates are easily conceived by way
of cyanide substitution of a 1º-halide, coupling of a Gilman reagent with
allyl bromide, or Grignard addition to ethylene oxide.
The last disconnection (green arrow) creates the desired carbon skeleton by
sequential alkylations of terminal alkynes (first acetylene and then
4-methyl-1-pentyne). Mercury catalyzed hydration of the symmetrical octyne
product generates the desired ketone. All the necessary reactants are
C4 or less, so the synthesis is accomplished in three steps (not
counting the formation of alkyne salts).
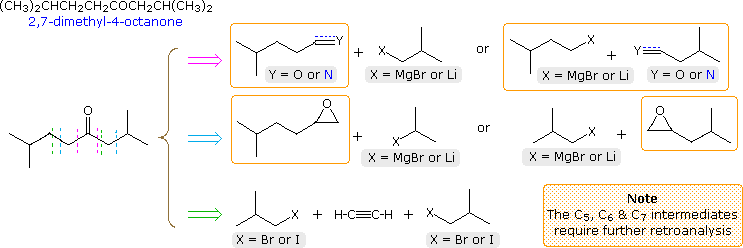
Three more first-stage analyses will be displayed above by clicking on the diagram. The first of these (red arrow) is a
two step sequence initiated by isobutyl magnesium bromide addition to
acetonitrile, followed by isobutyl bromide alkylation of the resulting
4-methyl-2-pentanone. Regioselective control might be a problem in the last
step. The second disconnection (orange arrow) suggests an α,
α'-dialkylation of acetone. Since acetone itself is prone to base-catalyzed
condensation, this might be difficult to accomplish directly. However, the
use of ethyl acetoacetate avoids this problem for the first step, and the
second alkylation is the same one proposed as part of the first
disconnection synthesis. Both of these sequences would provide efficient
routes to the target ketone.
Finally, the last disconnection is a four component assembly consisting of
two conjugate additions and a Grignard addition. This would most likely
result in a longer and lower yield procedure than the previous two.
Problem 3
A synthesis of 1,4,6--trimethylnaphthalene from para-xylene and
other starting compounds having no more than four contiguous carbon atoms
is required. Plausible transforms for the attachment of the second ring
carbons to para-xylene are Friedel-Craft
alkylation or acylation (acylation is usually better), nucleophilic attack
of an aryl metal
reagent derived from 2-bromo-para-xylene on carbonyl or epoxide
electrophiles, or possibly by cycloaddition to a aryne
intermediate. A palladium
catalyzed coupling reaction might also prove useful. Because of their
simplicity and broad scope, we shall consider only the first two
transforms.
The following diagram shows retrosynthetic analyses based on the
Friedel-Craft transform for both bond formations to the aromatic ring. Of
these, the first seems to offer the most efficient synthesis route,
consisting of Friedel-Craft acylation, Wolff-Kischner reduction, a second
Friedel-Craft acylation and methylation of a ketone enolate. In all cases
the substituted tetralone precursor of the desired naphthalene must be
reduced to an alcohol and dehydrated. The resulting dihydro naphthalene is
then aromatized by Pt catalyzed dehydrogenation, or mild oxidation by
heating with sulfur or selenium.
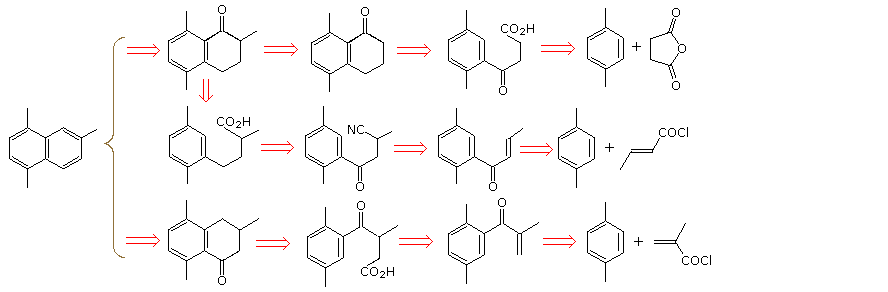
By clicking on the diagram, a new set of disconnections, starting from 2-bromo-para-xylene, will be displayed. A derived Gilman or lithium reagent is used for conjugate addition to an unsaturated carbonyl compound or ring opening of an epoxide. Further lengthening of the side chain is effected by cyanohydrin formation (top example), malonic ester alkylation (middle example), and Arndt-Eistert homologation (bottom example). The final steps must then parallel those used for the first examples.
Problem 4
A synthesis of 2-acetyl-2-methylbicyclo[2.2.2]octane from cyclohexene
and other starting compounds having no more than four contiguous carbon
atoms is required. The target molecule has two bridged six-membered carbon
rings, and cyclohexene is one of the starting materials. Whenever a
six-membered carbon ring must be formed, possible Diels-Alder transforms
should always be considered. For such a construction one needs a conjugated
diene and a dienophile. Cyclohexene might be considered a dienophile, but
acting as such would lead to a fused ring product, not a bridged ring
structure. Also, commonly used electron-rich dienes are not expected to
react well with an unstrained, electron-rich alkene.
If the role of cyclohexene is changed to that of a diene, these objections
are overcome. This alteration is easily managed by addition of bromine to
cyclohexene, followed by a double elimination, yielding
1,3-cyclohexadiene.
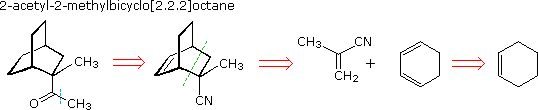
The possible use of cyclohexadiene in this synthesis is shown above. A Diels-Alder cycloaddition to a dienophilic double bond generates the desired bicyclooctane ring system, and the task is to identify a reasonable intermediate for this purpose. Among the many reactions that form ketones, the addition of a Grignard reagent to a nitrile is particularly efficient. If we choose this as the last step, the dienophile becomes 2-methylacrylonitrile, and the retrosynthetic path is complete. The isolated double bond produced by the cycloaddition is reduced by catalytic hydrogenation, so distinction between exo and endo-addition products is lost (the endo-adduct shown predominated).
Problem 5
A synthesis of 2-benzyl-3,3-dimethylcyclohexanone from benzene
derivatives having no more than seven carbons and other starting compounds
having no more than four contiguous carbon atoms is required. Since
conjugate addition of a methyl group to
2-benzyl-3-methyl-2-cyclohexen-1-one should proceed in good yield, this
unsaturated ketone provides a good alternative target, as shown. Once
again, the cyclohexane ring suggests a Diels-Alder transform. Three such
disconnections are depicted in the following diagram along with a possible
aldol cyclization (example 4). Diels-Alder approach 1 is the most
promising, since it features an electron-rich diene reacting with an
electron deficient dienophile. Chloroacrylonitrile is a useful surrogate to
ketene as a dienophile (ketene normally reacts by [2+2} cycloaddition).
Hydrolysis of the α-chloronitrile unit in the adduct converts it to a
carbonyl group. Unfortunately, the regioselectivity of this cycloaddition
is likely to be poor, with 5-benzyl-4-methyl-2-cyclohexen-1-one (orange box
bottom left) being formed in significant or possibly major amount. Also,
the diene, (3E)-3-methyl-5-phenyl-1,3-pentadiene, needed for this reaction
may be difficult to obtain as the desired stereoisomer (the Z-isomer will
be relatively unreactive because of steric hindrance in the cisoid
conformation).
Diels-Alder synthesis 2 does not have a regioselectivity problem, but the
reaction of an electron-rich diene with an electron-rich dienophile is
often sluggish and incomplete. Also the initial adduct has a methyl ether
where a carbonyl function is needed. The third Diels-Alder proposal in the
gray-shaded area has even more problems. As in reaction 2, electronic
factors make the cycloaddition poor, and the regioselectivity will likely
favor the wrong adduct (circled in orange). Even if the desired
3,3-dimethylcyclohexanone were obtained, benzylation at the desired
α-position (green) will have to compete with that at the less hindered
α'-position (magenta).
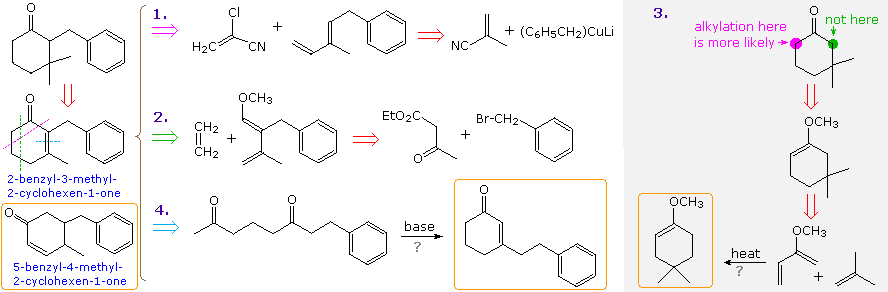
By clicking on the diagram, a new set of
disconnections will be displayed. The first of these (top line) is a cyclic
aldol transform similar to the last case discussed. Here, however, the
symmetry of the 1,5-diketone (after decarboxylation) permits only one
cyclohexenone product, 3-methyl-2-cyclohexen-1-one (drawn in the light gray
box). This key synthetic intermediate, known as a synthon, may lead
to the target molecule in two ways, depending on the order in which
conjugate addition and α-alkylation are conducted. Another useful concept,
revealed by the disconnections in the last two rows, is that benzene
derivatives may serve as precursors to cyclohexane compounds.
By clicking on the diagram a second time, the
reactions which may be used to achieve the proposed constructions will be
shown above. Note the use of a Birch
reduction in the second line. All three approaches should produce the
target compound, the most efficient arguably being the third.
Problem 6
A synthesis of all-cis-1,2,3,4-tetrakis(hydroxymethyl)cyclopentane from simple starting materials (six or fewer contiguous carbons) is required. Since carboxylic acids, esters, aldehydes and 1º-alcohols are easily interconverted, this target may be changed to the corresponding tetracarboxylic acid, as shown in the following diagram. Constructing the cyclopentane ring becomes a primary goal, and this may be done by condensation reactions (first two disconnections), cycloaddition (third disconnection) or by starting with a cyclopentane reagent (last example). Although there is precedent in known chemistry for all these approaches, some turn out to have serious flaws.
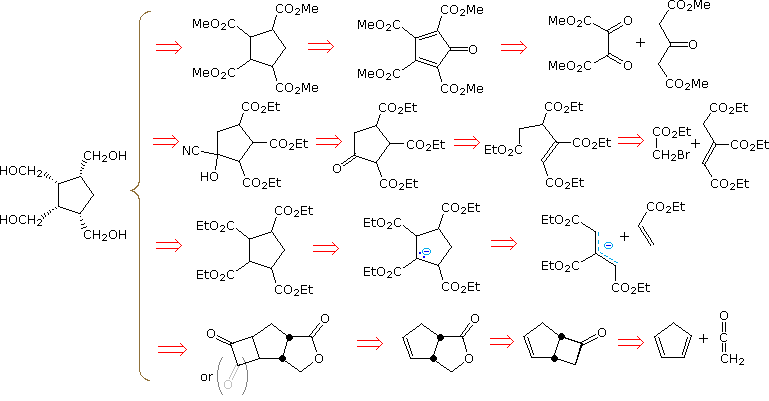
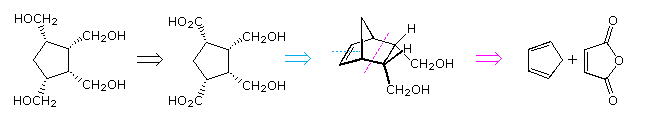
By clicking on the diagram, chemical reactions corresponding to each of the disconnection paths will be shown above. The first example, which takes advantage of symmetry, turns out to suffer from subsequent rapid Michael addition of a second acetonedicarboxylic acid moiety to the intermediate cyclopentadienone. This is, in fact, a general synthesis of bicyclo[3.3.0]octane-3,7-diones, known as the Weiss reaction. The second approach constructs the five-membered ring by a Dieckmann condensation of a tetra-carboxylic ester prepared from triethyl aconitate. Addition of the fourth carboxyl group by way of a cyanohydrin should be straightforward, but a mixture of stereoisomers will result, with the all-cis compound being a minor component. The cycloaddition proposed for the third approach is allowed by orbital symmetry, but only a few examples have been observed. Pursing this synthesis would be unwise, because it suffers from the same lack of stereoselectivity as the second case. Finally, The last approach, involving sequential [2+2] cycloaddition of ketenes to cyclopentadiene, is longer and has an inherent problem associated with the regioselectivity of the conventional Baeyer-Villiger oxidation. This problem may be overcome by using chiral catalysts (enzymes or transition metal complexes) with hydrogen peroxide, but a 50% conversion is the best that can be achieved and stereoselectivity may still be a problem.
A careful examination of the tetracarboxylic acid target reveals a possible precursor in which the cis carboxyl groups at C1 and C4 are masked by incorporation in a double bond. Such a bicyclo[2.2.1]heptene structure is readily achieved from 1,3-cyclopentadiene by way of a Diels-Alder reaction, as shown in the following retrosynthetic disconnection. With this as a guide, a simple three step synthesis may be proposed (shown by clicking on the diagram). The borohydride workup of the ozonolysis in the last step will convert aldehydes to 1º-alcohols.
Practice Problems
The following problems examine many aspects of organic synthesis. They are roughly organized by increasing difficulty.
Historical Background
One of the earliest, and perhaps most significant-although accidental-examples of synthesis was reported by Friederich Wöhler in 1828. In an experiment designed to prepare ammonium cyanate from silver cyanate, he heated the latter with ammonium chloride expecting the outcome shown below.
| AgOCN + NH4Cl | ——> | ? NH4OCN + AgCl |
The product Wöhler obtained did not correspond to the expected cyanate
salt, but was identified as urea, NH2CONH2, an
organic compound isolated from urine fifty years earlier. This result was
revolutionary in two respects. First it provided another example of
isomerism, in that ammonium cyanate, ammonium fulminate
(NH4O-N=C) and urea are all isomers, a novel concept for the
time. Second it cast doubt on the widely held doctrine of vitalism, which
maintained that all living organisms were endowed with a vital or life
force that rendered them and their component parts uniquely different from
ordinary "inorganic" matter. Thus, strongly heating organic substances such
as carbohydrates and proteins yielded water, ammonia and carbonaceous
solids (all inorganic), with loss of the vial essence. Wöhler's experiment
was acclaimed as the first conversion of an inorganic substance into an
organic compound.
Less than twenty years later, the German chemist Adolf Kolbe provided an
even more convincing synthesis of organic from inorganic substances. The
two equations written below outline his experiment. First, carbon
disulfide, obtained by reaction of carbon with sulfur, was converted to
carbon tetrachloride by heating with chlorine, and the simultaneous
pyrolysis of CCl4 yielded a mixture of products which included
tetrachloroethene, presumably formed from dichlorocarbene
(:CCl2). Treatment of tetrachloroethene with aqueous
chlorine (think HOCl) gave trichloroacetic acid, which Kolbe reduced
electrolytically to acetic acid. This ended the reign of vitalism as a
scientific theory.
| CS2 + Cl2 + heat | ——> | CCl4 + Cl2C=CCl2 + many other products |
| Cl2C=CCl2 + Cl2 & H2O | ——> | CCl3CO2H ——> CH3CO2H |
During the 1850's, the French chemist Pierre Berthelot synthesized
scores of simple organic compounds, ranging from ethanol to acetylene and
benzene, setting the stage for more ambitious attempts. Just as the
alchemists sought to transmute base metals into gold, early organic
chemists were drawn to the isolation or preparation of rare dyes, exotic
perfumes and unusual spices, often worth more than their weight in gold. A
notable example of this interest is William Perkin's attempt to synthesize
quinine.
Quinine, an important drug for the treatment of malaria, was available only
from the bark of the South American tree Cinchona officinolis, and
in the mid 1850's a decline in the native tree population had caused a
large rise in the price of the drug. Very little was known about the
compound, other than its molecular formula
C20H24N2O2. Nevertheless, in
the spring of 1856, William H. Perkin, a student (age 18) at
the Royal College of Chemistry in London, attempted its synthesis in his
home laboratory. Perkin reasoned that oxidation of a suitable 10-carbon
amine, such as allyl toluidene, C10H13N, might
generate quinine, as shown in the following equation.
| 2 C10H13N + 3 K2Cr2O7 | ——> | ? C20H24N2O2 + H2O |
This simple approach failed, and from our vantage point a century and a
half later it is easy to see why. Many thousands of isomers having the
molecular formula of quinine are possible, but only one unique
configuration of these 48 atoms constitutes a molecule of quinine. That
the atoms of allyltoluidine should, in the course of one reaction,
selectively reorganize and combine in this specific fashion is beyond all
reasonable probability.
Perkin's experiment was a failure only in the respect it did not yield
quinine, and his subsequent study of aromatic amine oxidations demonstrates
the value of persistence. From an impure sample of aniline he obtained a
purple dye he called aniline purple (also called mauve), which became the
cornerstone of the synthetic dyestuff industry in Europe and made a fortune
for its discoverer.
A total synthesis of quinine was achieved in 1944 by R. B. Woodward and W. E. Doering (Harvard), and improved
syntheses continue to be reported.