Dissolving Metal Reductions of π-Electron Systems
Reduction of alkynes and benzene rings by solutions of sodium or lithium in liquid ammonia have been described. Other reactive metals, such as zinc and magnesium have played a role in reductions of aldehydes and ketones (Clemmensen reduction), alkyl halides and vicinal-dihalides. The ability of certain metals to donate electrons to (reduce) electrophilic or unsaturated functional groups has proven useful in several reductive procedures. The facility with which various of these metals donate electrons is given by their reduction potentials. From these potentials the qualitative order of reducing power is: Li > K > Na > Mg > Al = Ti > Zn > Fe > Sn.
Reduction of Isolated Carbonyl Groups
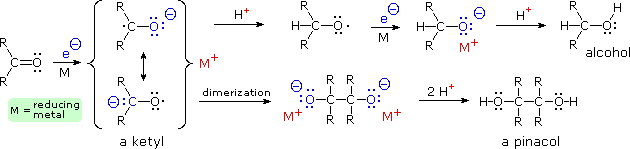
Lithium, sodium and potassium reduce ketones by a one-electron transfer that generates a radical anion known as a ketyl. Once such a reactive species is formed, it may react further by several modes, as described in the following diagram. If a proton source is present, the ketyl undergoes carbon protonation, and the resulting oxy radical adds another electron to generate an alkoxide salt. Alternatively, ketyls may dimerize to pinacol salts. Isolation of alcohol or pinacol products requires further protonation by acids at least as strong as water or ethanol. The H+ notation refers to any of several possible proton sources, including ammonia, alcohols and the ammonium cation (a strong acid in the liquid ammonia system). Benzophenone (diphenyl ketone) forms a deep blue ketyl which is stable in solvents that lack acidic hydrogens, such as hydrocarbons and ethers. It is widely used as an indicator of oxidizing or acidic impurities during the purification of such solvents.
The solvents used for alkali metal reductions include hydrocarbons, ethers and, most commonly, liquid ammonia. Alcohols may also be used, but usually as co-solvents, since they react vigorously with these metals. Examples of metal reductions of ketones to alcohols and pinacols (a dimeric diol) are shown below. In the first example, reduction of benzophenone in liquid ammonia gives both alcohol and pinacol products. The ketyl intermediate in this reaction is stabilized by phenyl substituents, and competitive carbon atom protonation and dimerization generate alkoxide salts that remain in solution until hydrolyzed prior to product isolation. In the second reaction, two isolated ketone functions are reduced to alcohols. The ketyl intermediates are not stabilized, and their rapid protonation is assured by the alcohol cosolvent. Conformational motion is restricted by the rigid polycyclic carbon framework of the substrate, and an interesting stereoselectivity is revealed: both alcohols are formed as the equatorial isomer. Aldehydes are not usually reduced in this manner, because they react with ammonia to form unreactive imine condensation products.
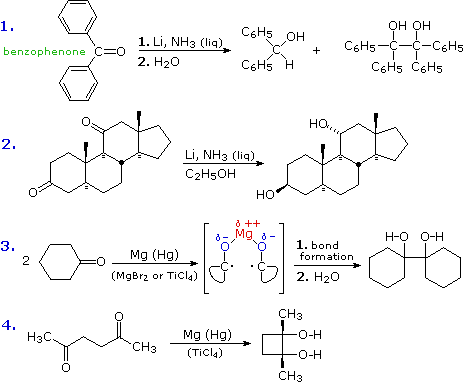
When pinacol products are desired, a less reactive metal having stronger (less ionic) C-O bonds is chosen for the reduction. Magnesium is often used, and best results have been achieved when the metal is activated by amalgamation (alloyed with mercury) and Lewis acids are present. Equations #3 & 4 (above) illustrate pinacol reduction. A di-positive cation may serve to hold two associated ketyl moieties close to each other so that bonding is facilitated (as shown in equation #3). Hydrolysis of metal alkoxides releases the product.
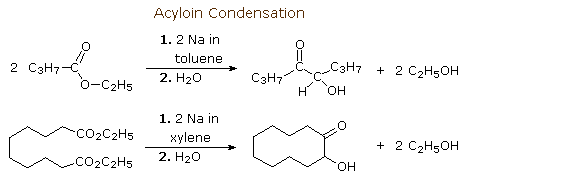
Ester functions undergo similar reductions on treatment with sodium. The most useful reaction of this kind is the acyloin condensation. To avoid protonation at carbon, this reaction is normally carried out in hydrocarbon solvents. The acyloin condensation creates alpha-hydroxy ketones. Two examples of this reaction are shown here. The second illustrates the usefulness of this reaction for constructing medium and large-sized rings. By clicking the "Show Mechanism" button a diagram for a possible mechanism for the acyloin condensation will be displayed. The reduction of alpha-diketones to acyloins, as shown on the second line, can be carried out independently.
Reductive Removal of α-Substituents
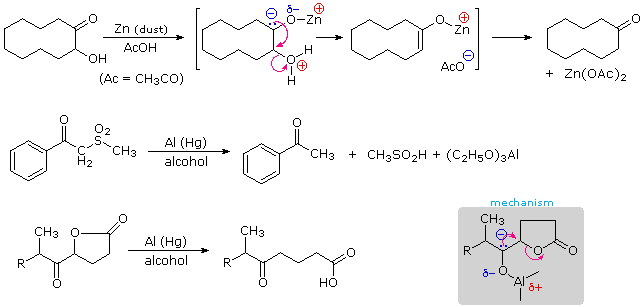
The partial negative charge on the carbon atom of a ketyl may serve to eliminate an electronegative substituent at an alpha-location. If further reduction is not desired, aluminum or zinc are often selected for this reductive elimination. The following examples illustrate three such transformations, the first being a useful conversion of acyloins to ketones.
Reduction of Conjugated π-Electron Systems
Two or more different functional groups are sometimes found together, and interaction of one upon another may lead to unexpected chemistry. The addition reactions of conjugated dienes are one example of this phenomenon. A similar situation occurs in conjugated enones, compounds in which a carbonyl group is bonded to a carbon-carbon double bond.
C=C–C=O |
(an α,β-unsaturated ketone or enone) |
Such functional combinations are often prepared by an aldol condensation, and are particularly useful as synthetic intermediates. Because the π-electron systems of the two functional groups are conjugated (the π-orbitals overlap in space), the radical anion formed by electron addition from a reducing metal is a resonance hybrid of six canonical structures. In addition to the two ketyl contributors described above, two structures having radical and nucleophilic character at the beta-carbon are shown in the following diagram, and two others in which the radical anion character is localized on the double bond are probably least important.
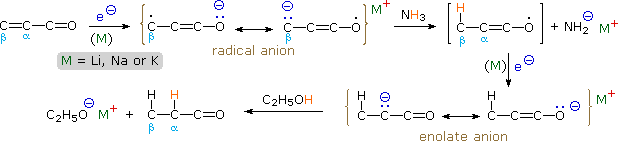
The usual fate of the extended ketyl described here is protonation (or other electrophilic bonding) at the beta-carbon atom. This creates an enoxy radical which immediately accepts an electron to form an enolate anion. Protonation or alkylation of this enolate species then gives a saturated ketone, which may be isolated or further reduced depending on the reaction conditions. Four examples of such reactions are shown below.
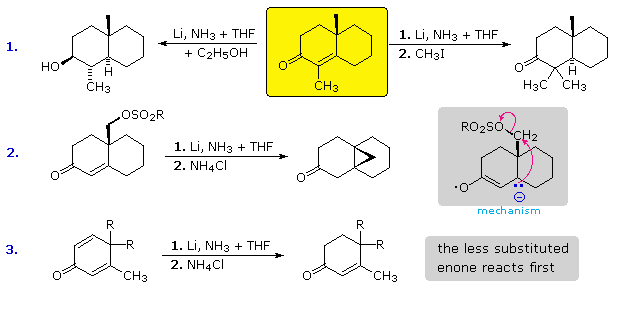
In example #1 the enone substrate is drawn in the yellow box. If the lithium reduction is carried
out in liquid ammonia without any acidic co-solvents, the enolate anion is stable and remains
unchanged until an electrophilic reagent such as methyl iodide is added. This is shown for the
reaction to the right. If an acidic cosolvent such as ethanol is present, the enolate anion is
protonated, and the resulting ketone is then reduced to an alcohol (reaction to the left).
Although the radical anion intermediate usually undergoes protonation at the beta-carbon, this is
not a fast reaction in liquid ammonia. Example #2 presents an interesting case in which
intramolecular alkylation of the beta-nucleophile occurs faster than protonation. Example #3 is a
case of cross-conjugation. The carbonyl group is conjugated with one or the other double
bond, but not both simultaneously. Two different radical anions may be formed by electron
addition, and these exist in equilibrium with each other. Protonation at a beta-carbon effectively
traps a radical anion as its related enolate anion, preventing any further interconversion. This
protonation is fastest at the less substituted site (upper enone), and if the resulting enolate
anion is not converted to its keto form by in situ protonation, it will not react further until
quenched by ammonium ion.
Conjugated dienes are also reduced by sodium or lithium solutions
in liquid ammonia. 1,3-Cyclohexadiene is reduced to cyclohexene, but the unconjugated 1,4-diene is
not. If a double bond is conjugated with a benzene ring, as in styrene, it is likewise reduced.