Reactions of Enolate-Like Species
Regioselectivity in Enolate Anion Formation and Reaction
The importance of enolate anions as synthetic intermediates is well established. Nevertheless,
problems remain concerning their selective formation and reaction. For example, aldehyde enolate
bases are likely to undergo the
aldol reaction during their
formation, and ketones like 2-heptanone have two different alpha-carbons, each capable of
enolization. The ambident nature of
enolate anions also enables electrophilic attack at both oxygen and carbon, but in most synthesis
applications bonding to carbon is desired. Finally, enolate anions may often be formed as
E/Z stereoisomers, and it has been shown
that reaction stereoselectivity, when new chiral centers are created, depends on the enolate
configuration.
The following diagram illustrates how the conditions under which enolate anion
formation is accomplished can influence the
regioselectivity of the reaction.
The two ketone substrates, 2-heptanone and 2-methylcyclohexanone, each have differently
substituted alpha-carbons. In each case, enolate anion mixtures are generated by reaction with a
strong 2°-amide base (LDA is the usual
choice). If the ketone is added to a cold THF solution of excess base, enolate anion formation is
fast and irreversible (procedure a). On the other hand, if a slight excess of ketone is
allowed to remain in solution, an equilibrium involving the ketone and the various enolate species
is established (procedure b). At equilibrium the more stable enolate anion will
predominate. The examples given in the diagram also report results from an equilibrating
preparation in which the lithium metal in LDA is replaced by potassium (procedure c).
Regioselective Formation of Enolate Anions
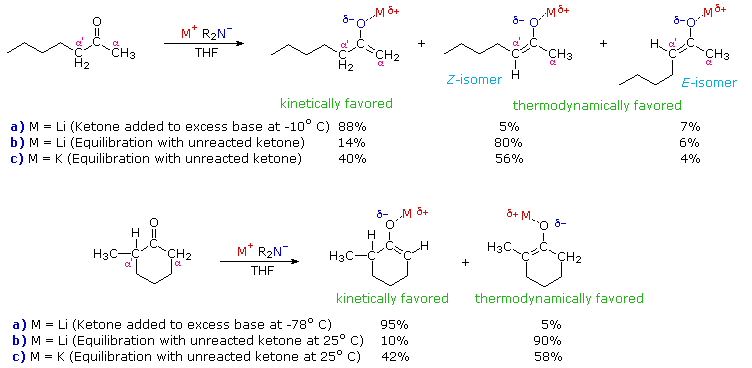
Several important principles are demonstrated here. First, if the enolate species has substantial double bond character, the more highly-substituted enolate double bond should predominate at equilibrium, as predicted from the stabilities of substituted alkenes. Since lithium-oxygen bonds are more covalent (have less ionic character) than potassium-oxygen bonds, the lithium enolate approximates an alkene more closely than the potassium enolate. Second, the greater ionic character of the potassium enolate places an increased negative charge on the alpha-carbon, a condition that is disfavored by alkyl group substitution. Indeed, the stability order of substituted carbanions is opposite to that of carbocations thanks to the electron donating character of alkyl groups relative to hydrogen. Finally, the rate of proton removal from an alpha-carbon site is decreased by alkyl substitution, probably reflecting a combination of steric hindrance (to bulky bases) and decreased carbanion stability. In both of the examples shown above, the conditions used in procedure (a) are typical of kinetically favored enolate formation, whereas those used in procedure (b) favor thermodynamic enolate formation. The comparative acidities provided by pKa values are derived from measurements made under equilibrating conditions, and therefore reflect thermodynamic acidity. Determinations of kinetic acidity require competitive isotope exchange experiments.
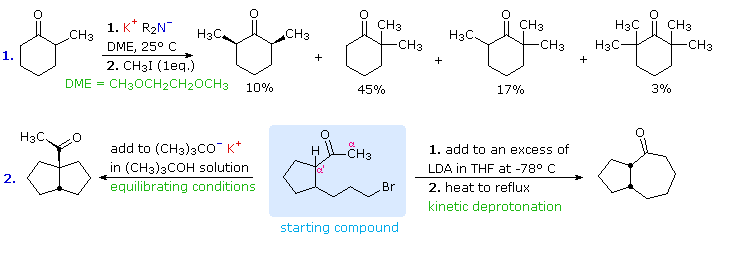
These principles influence the course of enolate alkylation reactions, as shown in the following
diagram. In the first case, 2-methylcyclohexanone is converted to a thermodynamic enolate mixture,
which is then reacted with methyl iodide. The major product is the expected
2,2-dimethylcyclohexanone (from the more stable enolate anion), but this is accompanied by di- and
trimethylated products together with about 20% unreacted starting material. The complexity of the
product mixture is due to acid-base proton transfer between alkylated products and unreacted
enolate anion. In other words, once a small amount (say 5%) of dimethylcyclohexanone is formed, it
finds itself in solution with a relatively high concentration of a strong base (the remaining
enolate anion) that can remove another alpha-proton, giving a new enolate anion that is further
methylated. If the kinetically favored lithium enolate (see the previous diagram) is used instead
of the equilibrium potassium enolates, 2,6-dimethylcyclohexanone is the chief product.
The
second reaction is an intramolecular alkylation that can occur in two different ways. If the
kinetically favored enolate (methyl proton removal) is formed at low temperature, it reacts
rapidly on warming to form a seven-membered ring. Alternatively, the weaker base, potassium
tert-butoxide (in the alcohol as solvent), generates an equilibrium mixture of enolates which
eventually react by intramolecular alkylation. The thermodynamically favored α'-enolate
predominates, and the resulting alkylation generates a five-membered ring.
Examples of Selective Enolate Alkylation
Another aspect of enolate anion alkylation, not yet addressed, is the possibility of electrophilic
bonding at oxygen. One example of such behavior will be displayed by clicking the "Toggle Reactions" button. Because of the substantial negative charge on the oxygen of ambident anions, it might
be expected that O-alkylation would be the rule rather than the exception. This, in fact, is true
when fully or extensively ionized enolate salts are reacted with strong electrophiles. Ionization
of enolates is facilitated by high dielectric solvents, such as DMSO and DMF (dimethylformamide),
especially for potassium and cesium cation salts. As shown in the lower part of the second
diagram, the negatively charged oxygens of DMSO cluster about a cation, providing substantial
solvation stabilization. No such solvation exists for the enolate anion, leaving it open to
reaction with an electrophile. Lithium enolates have significant covalent character in the
metal-oxygen bond, and this retards electrophile attack at oxygen.
Ether solvents such as THF
and DME (dimethoxyethane or glyme) are commonly used for alkylations because they are inert to
strong base and dissolve enolate salts more effectively than hydrocarbons. The difunctional ether
DME (dimethoxyethane) is especially effective at solvating cations; and this fact has led to the
preparation of cyclic polyethers, known as crown ethers, which are extraordinarily powerful
solvating agents. Crown ethers may be added to enolate salt solutions to enhance their ionization.
Indeed, the size of the crown ether can be tailored to fit the cation being used, providing
additional control over the course of enolate reactions.
The nomenclature of crown ethers
consists of two numbers. The first (larger) number designates the overall ring size. The second
number indicates the number of ether oxygens. A symmetrical arrangement of the oxygens in the ring
is assumed.
Preparation and Reactions of Silyl Enol Ethers
One way of producing selective enolate anion intermediates is to first trap and isolate them as silyl enol ethers. These relatively stable compounds may then be used to generate isomerically pure enolate anions, or in some cases as enolic nucleophiles in their own right. In the following diagram, the first reaction illustrates the formation of a mixture of silyl enol ethers under equilibrating conditions. If a higher proportion of the minor isomer is desired the kinetically favored lithium enolate can be prepared and quenched with trimethylsilyl chloride. In either case the silyl ether mixture may be separated by distillation. Once a pure silyl ether isomer is in hand, it may be used to generate the corresponding lithium enolate in the manner shown. Alkylation reactions of these enolates then produces pure regioisomeric products.
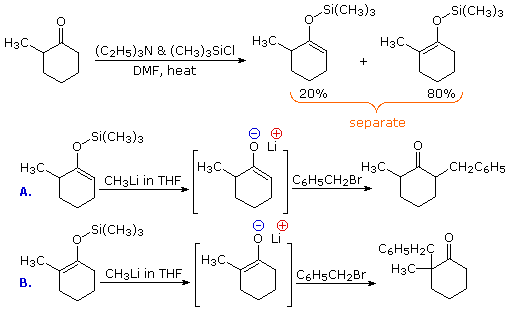
By clicking the "Toggle Reactions" button under the previous diagram, two examples of the direct use of silyl enol ethers will be displayed. Since the silyl ethers are not as reactive as enolate anions, the electrophiles with which they combine must be made more reactive. When carbonyl electrophiles are used, this can be accomplished by Lewis acid catalysts, as shown.
Enamines as Enolate Anion Surrogates
The formation of enamines by reaction of 2°-amines with aldehydes or ketones
has been described. The double bond
of the enamine transmits the nucleophilic character of the nitrogen to the alpha-carbon, in a
vinylagous fashion. Because of the resulting
ambident nucleophilicity of the enamines, reactions with electrophiles may take place at either
nitrogen or carbon. Enamines derived from aldehydes are usually alkylated on nitrogen, an
undesirable course for most synthetic applications. Ketones give significant C-alkylation, the
thermodynamically favored course, as first demonstrated by
G. Stork
(Columbia). The iminium ion created by C-alkylation cannot react further, and is easily hydrolyzed
to the alkylated ketone. This is particularly useful if dialkylation products are to be avoided.
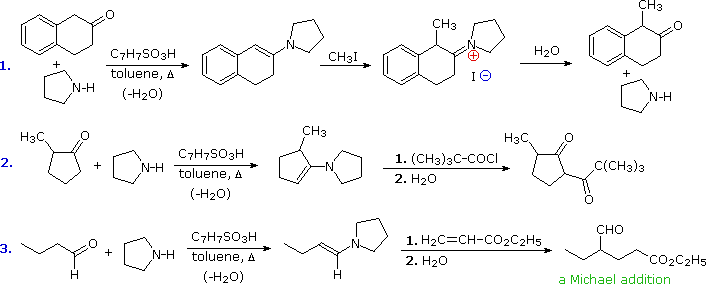
Thus, in the first example, direct methylation of the enolate anion from this ketone gives
significant amounts of the dimethyl product, due to enolate proton exchange.
As shown, the enamine route gives only mono-methylated product.
The second example
demonstrates that enamines may be acylated as well as alkylated. In fact, the reversible nature of
acylation removes the problem of competing N-acylation. This case also illustrates the general
tendency to form the least substituted enamine when two different alpha-sites are present.
Conjugation of the non-bonding electron pair on nitrogen with the pi-electrons of the double bond
forces the alkyl substituents on nitrogen to lie in the same plane as the double bond (see the
resonance equation displayed by the "Toggle Mechanism" button). As a result substitution of
the double bond leads to increased steric hindrance with the nitrogen substituents. The
five-membered cyclic 2°-amine pyrrolidine is widely used for enamine reactions, in part because
this steric hindrance is minimized.
Examples of Enamine Reactions
As noted, N-alkylation of enamines is common for aldehydes and some ketones. Michael addition reactions avoid this problem thanks to their reversibility. The third example shows such a reaction, and the "Toggle Mechanism" button displays a possible mechanism. The C-alkylation intermediate is thermodynamically more stable than the N-alkylation species, so it predominates at equilibrium. Both the charges in this intermediate are stabilized by delocalization, and hydrolysis rapidly converts it to the aldehyde-ester product. An interesting alternative is ring closure to a neutral enol ether compound (shown in the blue shaded box) which would also be hydrolyzed to the same product.
Imine and Hydrazone Anions
Still another way of circumventing some of the undesired aspects of enolate anion chemistry is to replace the oxygen of an aldehyde or ketone substrate with a 1°-amino group, in other words, to convert the carbonyl function to an imine. Imine derivatives are relatively easy to prepare, starting with an aldehyde or ketone and a 1°-amine or hydrazine derivative. The resulting C=N function does not activate alpha-C-H groups as effectively as a carbonyl function, but very strong bases such as LDA, alkyl lithiums and Grignard reagents will convert imines to their enamide conjugate bases quantitatively. This general reaction is shown in the green shaded box below.
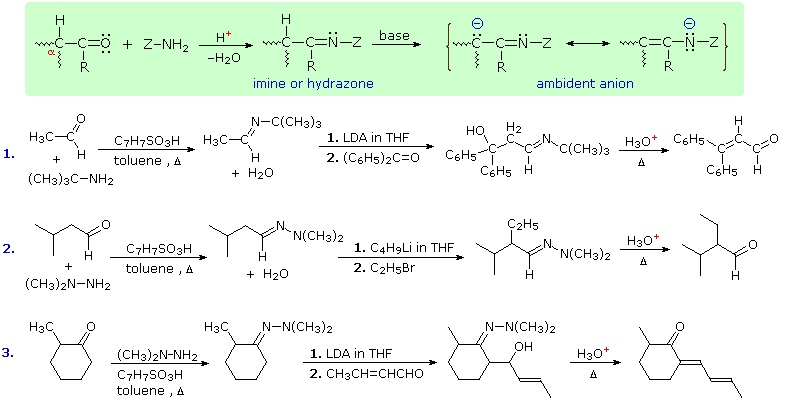
Three illustrations of the use of enamide bases in synthesis are displayed above. The first two examples use aldehyde derivatives, and if we were to attempt these reactions with the aldehyde enolate anion itself, aldol dimerization would result. The C=N function of imines is a poor acceptor of nucleophiles, so it does not assume such a role in aldol-like reactions. The third reaction is an aldol condensation in which a ketone serves as the donor. If cuprous salts are introduced before the unsaturated aldehyde is added to the enamide solution, conjugate addition takes place in preference to the 1,2-aldol addition.