Stereoelectronic Effects
Factors that influence the properties or reactivity of molecular species as a consequence of the
spatial orientation of filled or unfilled orbitals are termed stereoelectronic. In many
cases, electron pairs in atomic or molecular orbitals that are involved in the making or breaking
of bonds have an optimal geometrical alignment that is critical for a reaction to occur. This
alignment provides the best overall bonding of participating species during the course of a
reaction, and reflects the fact that transition states having the greatest bonding energy have
lower potential energies than transition states with less bonding.
Two relatively simple
examples of stereoelectronic effects are found in SN2 and E2 reactions. The mechanisms
of these reactions were described earlier in our discussion of
alkyl halide chemistry, but a
more extensive examination of these common transformations will provide a helpful introduction to
this subject.
SN2 Reactions
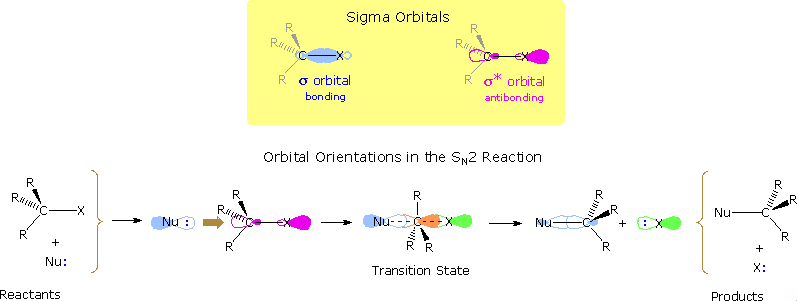
Nucleophilic substitution reactions of alkyl halides take place by a continuum of mechanisms, defined by SN2 behavior at one extreme and SN1 behavior at the other. The essential characteristics of the SN2 process include inversion of configuration at the reaction site, sensitivity to steric hindrance in the alkyl group, and second order kinetic behavior. The orbital interactions that take place in a successful SN2 reaction are shown in the following diagram. The bonding and antibonding sigma molecular orbitals composing the C-X bond are drawn in the yellow box at the top of the diagram. As a nucleophile approaches the rear side of the carbon, the orbital containing the non-bonded electron pair (light blue) begins to overlap with the empty antibonding C-X sigma orbital (pink colored), shown on the left of the bottom line. As electrons occupy this antibonding orbital, the C-X bond is weakened. In the transition state partial bonds exists between the carbon and both the nucleophile and the halogen (the carbon is essentially sp2 hybridized with the orange colored p-orbital providing the partial bonding). Finally, a full C-Nu sigma bond develops and the halogen leaves as an anion.
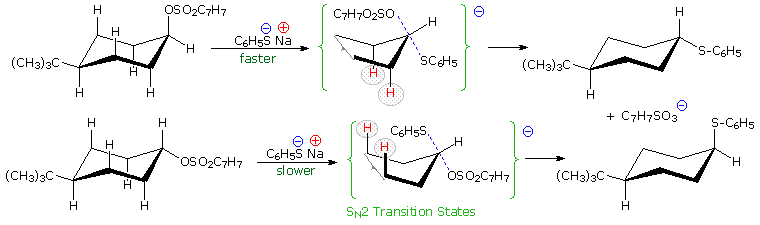
Other substitution reactions also proceed by the SN2 pathway, as illustrated by the stereoisomeric cyclohexyl tosylates in the following diagram. The bulky tert-butyl group on C-4 serves to lock the chair conformation in the configuration shown, so one isomer has an equatorial tosylate function, and the other an axial tosylate. On reaction with sodium thiophenolate, both isomers undergo bimolecular substitution with inversion, the axial isomer reacting about thirty times faster than its equatorial epimer. Steric hindrance to rear side nucleophile approach by the red colored hydrogen atoms is present for each isomer, but the relief of steric crowding of the axial tosylate leaving group helps to facilitate substitution of the that isomer.
SN2 reactions may be intermolecular, as in these examples, or intramolecular. By
clicking on the above equations, an example of two sequential substitutions, the first
intermolecular and the second intramolecular, will be displayed. Note that the intramolecular
substitution shown here proceeds via the same orbital alignment described above. The reacting
moieties in intramolecular reactions are incorporated in the same molecule; consequently, such
reactions exhibit first order kinetic behavior instead of the usual second order kinetics. Because
enolate alkylation reactions are irreversible, the strained four-membered ring product is stable under the reaction conditions.
In general, intramolecular forms of bimolecular reactions are faster than their intermolecular
counterparts, since the reactive sites are held close together (their relative concentrations are as high as
possible). The rapid
lactonization of gamma and delta-hydroxy acids
compared with corresponding intermolecular esterifications is another example of this principle,
which is associated with a favorable
entropy change. Note, however, that
ring size has a profound influence on the intra- vs. intermolecular
dichotomy.
Maximum overlap of the electron orbital of the nucleophile with the antibonding sigma orbital of
the carbon substituent requires an approach from the rear side of the carbon, ideally 180° from
the leaving group. The importance of this orientation, which is shown in previous diagrams, was
made clear by an experiment conducted by
A. Eschenmoser
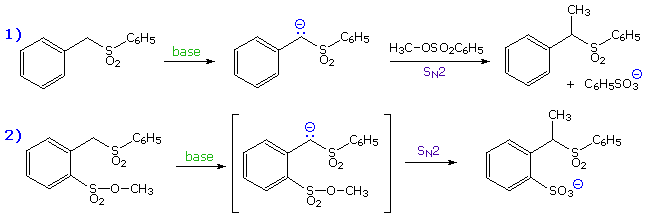
over 25 years ago. The results of Eschenmoser's experiment are presented in the following
illustration. Initially, two equations are written. The first describes the intermolecular
methylation of a sulfone anion by methyl tosylate, a typical SN2 reaction. The second
is a very similar reaction which many would consider to be intramolecular, since a methyl
sulfonate moiety is positioned ortho to the sulfone function.
By clicking on these equations,
evidence against the intramolecular mechanism will be displayed. Although intramolecular reactions
are often favored, this case requires a significant departure from the 180° orbital alignment
required by an authentic SN2 reaction. Since this reaction was found to be second order
and is not faster than the simple intermolecular analog (reaction 1 on the preceding
slide), it cannot be intramolecular.
E2 Reactions
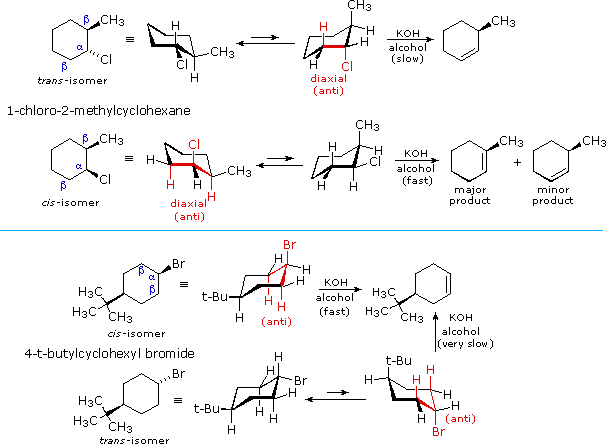
Elimination of vicinal groups, usually called 1,2- or beta-elimination, is the most common type of elimination reaction. Examples of some typical cyclohexyl halide eliminations are given in the following illustrations. From the location of the double bond in the elimination products, and the relative rates of elimination, it is clear that a diaxial orientation of the eliminating atoms or groups (colored red) is necessary for optimum reaction.
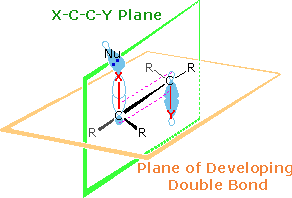
Two views of the characteristic orbital orientations of this anti-elimination are drawn on
the right. Two planes are defined in the upper drawing. The light green plane defines the coplanar
nature of the leaving groups (X & Y) and the carbons to which they are bonded. The light
orange plane identifies the plane of the final double bond. Together with the lower drawing, the
initial display shows the overlap of participating bonding orbitals. The corresponding antibonding
orbitals of the C-X and C-Y bonds will appear by clicking on the upper display. This orbital
alignment defines the stereoelectronic character of these elimination reactions. Note that the
"anti" descriptor for this configuration refers to the
antarafacial relationship of X and
Y with respect to the light orange plane. Also, the E2 designation refers to the second order
kinetics observed for these reactions.
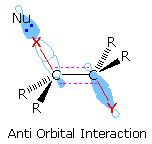
Although anti-eliminations are favored when possible,
syn-elimination is observed in some cases. By clicking on the lower display, it will be replaced
by a similar orbital drawing for syn elimination. In both syn and anti-transition states the
X-C-C-Y bonds are coplanar, an alignment that allows facile conversion of two sigma bonds (colored
red) to a pi-bond. Because of the eclipsed configuration of the syn-transition state, it is less
stable than a corresponding anti-transition state. Syn-elimination is sometimes observed when Y is
an ionic species, such as (CH3)3N(+); but it usually occurs when
an anti configuration is unstable or not possible, as in the following example. Isotopic
substitution confirms that this base-catalyzed beta-elimination takes place by a syn mechanism.
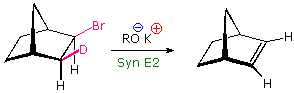
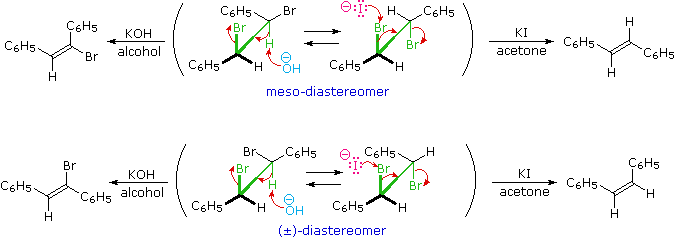
The anti-configurational relationship of the leaving groups in E2 reactions is relatively easy to see in the cyclohexyl compounds shown above, thanks to the chair conformations of these substrates. If this stereoelectronic feature is general for all E2 reactions, it should also hold for eliminations of acyclic compounds. The following examples illustrate this behavior. Conformational mobility about the central C-C bond allows each stereoisomer to adopt an anti alignment of leaving groups (colored green), and this is reflected in the products.
Trans-Addition to Alkenes
Addition reactions to carbon-carbon double bonds show a parallel stereoselectivity to the elimination reactions. Both syn and anti additions are observed, as noted in an earlier chapter. Indeed, the trans-addition of halogen reagents provides good examples of the importance of stereoelectronic control, even in the presence of opposing steric factors. The addition of bromine to 4-tert-butylcyclohexene, shown here, is a simple case. Assuming trans-addition, two stereoisomeric products are possible. Remarkably, the product having two axial bromines is favored over its diequatorial isomer. In order to maintain maximum bonding throughout the addition, carbon-bromine bonds must be aligned perpendicular to the plane of the double bond. This diaxial orientation mirrors the stereoelectronic demands of the E2 eliminations discussed above, and may be realized in two ways.
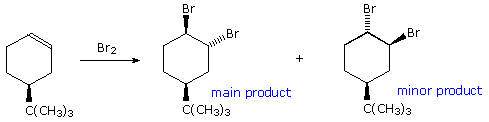
By clicking on the equation, the first and most favored addition will appear above. A half-chair
conformation for the substituted cyclohexene is drawn on the left. The large t-butyl substituent
occupies an equatorial orientation and serves to hold the conformer as shown. Trans-addition in
the manner depicted leads directly to the diaxial product. Clicking on the drawing a second time
displays the alternate trans addition. In order to maintain good orbital alignment throughout this
reaction, the cyclohexane ring must adopt a higher energy twist-boat conformation. This on
relaxing to a chair gives the diequatorial product. As a result, the first addition has a lower
activation energy than the second.
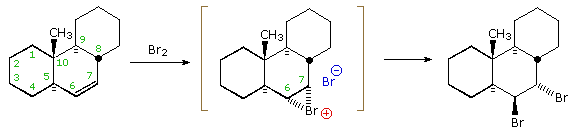
The attentive reader will recall that trans-addition of
bromine and other halogen reagents was attributed to nucleophilic ring opening of a
cyclic halonium ion intermediate. It
is instructive to consider this aspect of the stereoelectronic factor using a somewhat more rigid
alkene substrate. This is shown in the following illustration. The trans-fused six-membered rings
hold the central cyclohexene ring in a tight configurational grip. Chair-chair interconversions
are not possible, and even twist-boat conformers are very strained. The initial electrophilic
attack of bromine takes place from the underside of the molecule, due to the steric hindrance of
the axial methyl group. The resulting bromonium ion is written in brackets, and it is apparent
that nucleophilic attack from the top would be less hindered at C-7 than at C-6 (the methyl group
again). By clicking on the equation, a conformational drawing of this reaction will be displayed.
The two possible sites of attack are designated by green (C-6) and red (C-7) arrows. Although
bromide ion attack at C-6 is more hindered, it is favored by the diaxial stereoelectronic factor,
and this is the initially observed product. Subsequent rearrangement of this diaxial dibromide to
its diequatorial isomer takes place slowly.
Stereoelectronic control of epoxide ring opening follows the same principles.
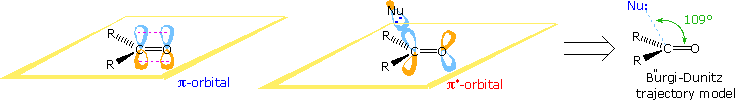
Addition of Nucleophiles to Carbonyl Groups
Stereoelectronic factors have been shown to influence the addition of nucleophilic reagents to carbonyl groups, particularly for aldehydes and ketones. An outline of this stereoelectronic effect is provided in the following diagram. The essential molecular orbitals are drawn to the left of the diagram, and the initial bonding with a nucleophile is believed to take place with the empty antibonding pi-orbital. This would explain the favored bonding alignment, known as the Bürgi-Dunitz trajectory.
Many carbonyl addition reactions have been analyzed by a combination of steric and stereoelectronic factors. In the following illustration, two hydride reductions of methyl ketones are shown. Since each ketone has an existing stereogenic site, and since the reduction creates a new chiral center, diastereomeric products are possible. These may be designated in several ways, but the syn-anti notation is generally preferred. When the existing stereogenic center is located next to the carbonyl group, as in the upper equation, it may influence the proportion of product diastereomers. Diastereoselectivity of this kind is sometimes called asymmetric induction. This influence is, of course, the same for both an enantiomerically pure or a racemic reactant. If, however, the stereogenic center is far away from the carbonyl group, it has a negligible influence on the reduction, and a 50:50 mixture of diastereomers is produced (lower example).
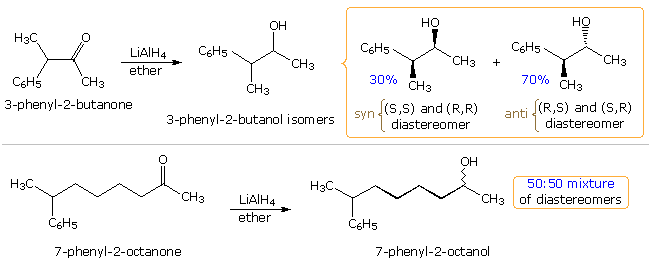
A number of models have been proposed to explain the diastereoselectivity of the first reduction.
Many conformations about the alpha-C-CO bond may be written, and the challenge is to pick one that
accounts for the observed selectivity. By clicking on the drawing, three of these will be
displayed. For general use, the substituents on the chiral center adjacent to the carbonyl group
are labeled L (large), M (medium) and S (small) to reflect their size. In
each model the sterically preferred Bürgi-Dunitz approach is shown by a pink arrow, and each
predicts the correct configuration of the favored diastereomer. The earliest rationalization was
offered by
D. Cram
and is shown by the model on the left. A more favored conformation was chosen by G. Karabatsos, as
shown by the center model. The most recent model (on the right) is that proposed by H. Felkin.
These models all require classification of S, M & L substituents, occasionally a tricky
process, and assume a reactant-like transition state for the reduction. Even so, some bonding of
hydride to the carbonyl carbon must take place in the transition state, accompanied by
corresponding small structural changes. Plausible structures for these transition states are given
in the orange box. An important point needs to be made here.
The ratio of products obtained from a a group of equilibrating conformers is determined by
transition state energies, not conformer concentrations.
This generally useful rule is called
The Curtin Hammett Principle.
A brief analysis of these models is instructive. The Cram model makes use of an unfavorable conformer (R & L are eclipsed). Although this strain is slightly relieved in the transition state, the oxygen and associated metals is moving toward the M group, resulting in increased crowding. The Karabatsos model starts with a favorable conformer, but the nucleophile trajectory nearly eclipses the C-S bond (~20° dihedral angle). The oxygen shift in the transition state relieves eclipsing strain with M, benefiting the transition state energy. Finally, the Felkin-Ahn model seems to offer the best rationalization. The nucleophile trajectory is roughly 40° away from eclipsing the C-S bond, and both the oxygen and the R group undergo favorable shifts in the transition state.
Cyclic ketones have fewer low energy conformations than do acyclic ketones, and diastereoselectivity in nucleophilic addition reactions is often observed. An analysis similar to that used above for the acyclic compounds has proven useful in explaining and predicting the outcome of such reactions. By clicking on the drawing a second time, the hydride reduction of 2-methylcyclopentanone will be displayed. The trans isomer is favored by a 3:1 ratio. Again, steric hindrance to hydride approach on the Bürgi-Dunitz trajectory rationalizes this finding.
For two additional examples of stereoelectronic effects in conformational equilibria Click Here.
Stereoelectronic Modulation of Cyclization
The formation of rings from suitably substituted molecular chains is a common requirement in synthesis. In general terms this transformation takes place by the bonding together of two reactive functional groups located at the ends of a chain. In the following diagram these are represented by an electrophilic group (E), and a nucleophilic group (Nu). Two possible modes of reaction are outlined here. In order to form a ring, an intramolecular reaction must take place. However, any significant quantity of reactant contains approximately 1020 molecules, and these may react with each other in an intermolecular manner to give dimeric and larger products. Controlling the course of such a reacting system is not a trivial matter, and many factors influence the outcome. Among these, the thermodynamic functions enthalpy and entropy are especially important.
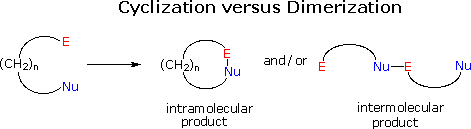
The enthalpy factor is associated with ring strain, which includes angle and eclipsing
strain. Small three and four-membered rings are
destabilized thermodynamically by such
strain, and as a rule cannot be prepared by reversible bond formation. Five and six-membered rings
are less strained, and are often created by reversible bond formation, as in the cases of
lactonization of hydroxy acids and
Dieckmann condensations. Medium sized rings (8 to 11 atoms in size) are generally destabilized by
transannular crowding (steric hindrance
by groups on opposite sides of the ring).
The entropy factor is related to the concentration
of chain conformations in which the ends to be joined are close together. For a three carbon chain
this concentration is nearly 100%, but it drops off rapidly as the length of the chain increases.
The number of different conformers a chain may assume increases with the length of the chain, and
the proportion of conformers having proximate ends decreases. These thermodynamic factors interact
in opposition to each other. Entropy favors small ring formation, even though this is
energetically (enthalpically) unfavorable. Irreversible bond forming reactions, such as enolate
alkylations, are used to achieve this outcome. Five and six-membered ring
formation is favored by both factors, and may be achieved by both reversible and irreversible bond
formation. The often cited principle that
intramolecular reactions are favored over intermolecular reactions has its origin here.
Medium and large sized rings, on the other hand, are difficult to make due to the low probability
of suitable conformers in the reaction mixture. Intermolecular dimerization and polymerization are
usually observed unless the reactions are conducted at high dilution. Remember, the
rate of a bimolecular reaction, such
as dimerization is proportional to the square of reactant concentration (2nd order); whereas, the
unimolecular cyclization reaction has a rate proportional to the first power of reactant
concentration (1st order). Thus, at very low reactant concentration, cyclization occurs faster
than dimerization.
Because of the structural constraints in a cyclization transition state, stereoelectronic influences may be anticipated. One example is shown by the following two equations. Conjugate addition of amines to unsaturated carbonyl reactants is usually rapid and reversible, as demonstrated by the first equation. At first glance, the reactant in the second equation might be expected to undergo an intramolecular reaction of the same kind, yielding a five-membered heterocyclic ester (drawn in the red box). In practice, however, the reaction is slower and takes a different course, producing a five-membered lactam by direct acylation of the amine by the ester.
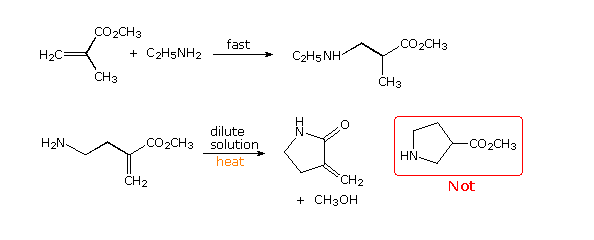
To explain this unexpected change in behavior, we must examine the orbital trajectories permitted in the two transition states (1,4- versus 1,2-addition). By clicking on the drawing a representation of these transition states will be displayed. Neither achieves a perfect Bürgi-Dunitz approach of the nitrogen nucleophile to a double bond, but the 1,2-addition comes much closer. Furthermore, the best 1,4-addition trajectory is unable to occur in a plane orthogonal to the plane of the double bond; whereas, the 1,2 addition is again much closer to the ideal orientation. For a display of 1,4- versus 1,2-addition in this example Click Here.
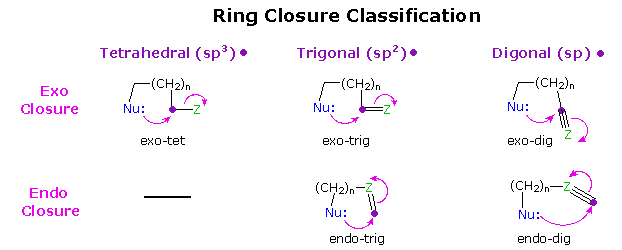
The Baldwin Rules
General stereoelectronic constraints on cyclization reactions were recognized by J. Baldwin, and
summarized in a qualitative model now referred to as the Baldwin Rules. The following
diagram illustrates the five major cyclization modes treated by Baldwin. In each case a
nucleophilic moiety bonds to an electrophilic site (usually a carbon atom), designated by a purple
dot. The flow of electrons during bond formation may be encompassed by the developing ring (an
endo cyclization), or may shift away from the ring (an exo cyclization). Furthermore, the
electrophilic site may have different hybridization states. The terminology used to identify a
given ring formation consists of three parts: the ring size, given by n + 3 in the diagram (where
n=0 or an integer), the exo / endo feature, and the hybridization. Since it is unlikely that
endo-tet reactions will be useful for making rings, such reactions are not included here.
However, the possibility of a six-membered cyclic analog of this kind was
discussed earlier.
Using this terminology, the previous cyclization of
the amino ester to a lactam is a 5-exo-trig process (the double bond is the carbonyl
group). The alternative ring closure by conjugate addition is 5-endo-trig, with respect to
the carbon-carbon double bond, even though the conjugative shift of electrons to the ester
carbonyl is exocyclic.
The stereoelectronically favored trajectories for nucleophilic bonding to carbon atoms of different hybridization states are summarized below. The tetrahedral and trigonal cases have already been described. It is clear that these trajectories represent a "least motion" transition state in which bonding is kept to a maximum throughout the reaction. By applying these trajectories to the five cyclization categories noted above, Baldwin was able to discern the effect of ring size on an idealized transition state. In some cases the constraints of a given ring size perturbed the transition state to such a degree that reaction seemed improbable. Such cases were called unfavorable.
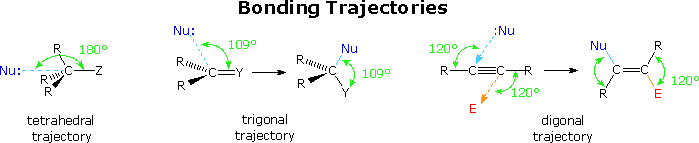
The following table summarizes Baldwin's conclusions. The 3-endo-trig, 4-endo-trig and 5-endo-trig
combinations, together with the 3-exo-dig and 4-exo-dig possibilities (all colored pink in the
table) were classified as "unfavorable". All the other combinations were classified "favorable",
and are colored light green in the table.
Remember, this evaluation is based primarily on
stereoelectronic factors. So called favorable cyclizations may fail for other reasons, and
sometimes an unfavorable cyclization may take place when alternative reactions are even less
favored.
Ring Closure Categories
| Tetrahedral Closure | 3-exo-tet | 4-exo-tet | 5-exo-tet | 6-exo-tet | 7-exo-tet |
|---|---|---|---|---|---|
| Trigonal Closure | 3-exo-trig | 4-exo-trig | 5-exo-trig | 6-exo-trig | 7-exo-trig |
| 3-endo-trig | 4-endo-trig | 5-endo-trig | 6-endo-trig | 7-endo-trig | |
| Digonal Closure | 3-exo-dig | 4-exo-dig | 5-exo-dig | 6-exo-dig | 7-exo-dig |
| 3-endo-dig | 4-endo-dig | 5-endo-dig | 6-endo-dig | 7-endo-dig | |
| Unfavorable combinations are colored light red | |||||
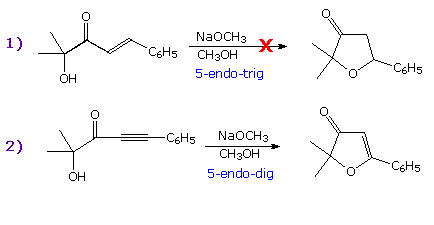
In the examples on the right the first equation describes a 5-endo-trig cyclization that
fails to take place. The second equation shows a successful 5-endo-dig reaction, similar in many
respects to the first.
When an enolate anion serves as the nucleophilic agent in a ring closure, a modification of the
Baldwin rules should be used. These rules will be shown on the right by clicking on the drawing.
An example comparing a 5-(enolexo)-exo-tet closure with an isomeric 5-(enolendo)-exo-tet option
will then replace the modified rules by a second click on the drawing. A third click on the
drawing will display a similar 6-(enolendo)-exo-trig example.
Rationalizing Conformational Preferences
The following diagram shows two conformational equilibria in which one of the isomers is favored.
Esters generally prefer to adopt a s-Z conformation (sometimes termed s-trans), as shown in the
first example. This notation, which refers to the alkoxy sigma-bond to the carbonyl carbon,
corresponds to that used for
diene conformations. Lactones
incorporated in seven membered and smaller rings are forced into an s-E conformation, and
consequently are more reactive.
In the second example, tri-tert-butyl
hexahydro-(1,3,6)-triazine exists predominantly in a configuration having one axial tert-butyl
group. Explaining these structural preferences provides an instructive review of stereoelectronic
factors.
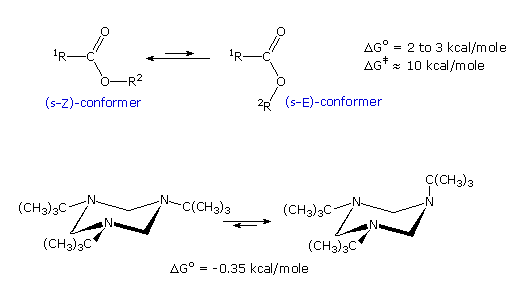
The ester equilibrium is most simply analyzed by considering electron and dipole interactions. The
latter will be displayed by clicking on the diagram. In the s-E conformer the carbonyl group
dipole is repelled by the similarly oriented ether oxygen dipole, whereas these dipoles have
opposite directions in the s-Z conformer. Also, the non-bonded electron pairs of the two oxygens
are closer together in the s-E conformer than in the s-Z conformer. A similar analysis of the
second example shows a parallel alignment of all three amine dipoles in the all equatorial
conformer, and a less repulsive arrangement in the axial conformer.
A thorough evaluation of
these cases should also examine the interaction of a non-bonding electron pair with neighboring
(vicinal) antibonding orbitals that may function as electron acceptors. This analysis of the ester
equilibrium will be shown by clicking on the diagram a second time. The upper part of the diagram
illustrates the p-pi conjugation of one non-bonding electron pair with the carbonyl group. This
conjugation explains the large energy barrier for interconversion of the E & Z conformers. The
remaining electron pair occupies a sp2 orbital (colored pink), and this is the donor
pair that may have a bonding interaction with an acceptor orbital (an
anomeric effect). Only the
Z-conformer provides this stabilizing interaction, which involves the antibonding C-O orbital
(light gray) of the carbonyl sigma bond (colored red). A similar donor-acceptor interaction
provides additional stabilization for the axial-tert-butyl conformer in the second example, as
illustrated by clicking on the diagram a third time.
The Anomeric Effect
The anomeric effect was first recognized in carbohydrate chemistry, but applies generally to all organic compounds.