Molecular Structure and Acidity
Equilibrium Acidity: pKa Acidities of Some Common Hydrides
The relative acidities of different acids are commonly measured and cited as pKa values, relative to a standard solvent base, often water. These numbers reflect the equilibrium acidities of the acids. An astounding range of acidities is displayed by even rather simple compounds. The table below lists some elemental hydrides from groups 4 through 7 of the periodic table. The pKa's determined (or in some cases estimated) for these compounds are shown beneath the formulas. Approximate values for higher members of group 4 and 5 hydrides (e.g. silane and phosphine) have not been reported. Note that these logarithmic numbers encompass nearly sixty powers of ten. This is a greater span than that encompassed by distance measurements starting from the radius of a hydrogen atom and extending to the diameter of the known universe.
| 4 | 5 | 6 | 7 |
|---|---|---|---|
|
CH3-H ca. 50 |
NH2-H 34 |
HO-H 15.74 |
F-H 3.2 |
|
HS-H 6.97 (pK1) |
Cl-H -3 |
||
|
HSe-H 3.8 (pK1) |
Br-H -6 |
||
|
HTe-H 2.6 (pK1) |
I-H -7 |
Why do these relatively simple compounds differ in acid strength so markedly? Two factors may be discerned:
- First, the compounds in the top row clearly show the importance of electronegativity. All the heavier elements have greater electronegativities than hydrogen, with carbon being the least different. The ionic character of these covalent bonds is such that hydrogen carries a partial positive charge, and the heavier atom a corresponding negative charge. The greatest charge separation is in H-F, where the electronegativity difference is nearly 2. Removal of a proton is facilitated by this charge separation. The covalent bond energies do not correlate inversely with acid strength, as one might have expected, since the two strongest acids have the strongest bonds (H–O 111 kcal/mol & H–F 135 kcal/mol). Finally, the heavy atoms in the top row have similar sizes, the covalent radii being 0.75 ±0.02 Å. The importance of this fact will become apparent in the next discussion.
- Second, the compounds in the columns representing periodic groups 6 and 7 show an increase in acidity moving from the top to the bottom. This is opposite to the electronegativity change, and is best attributed to an increase in heavy atom size. When an acid transfers a proton to a base, the remaining residue (the conjugate base) must carry a negative charge. Ignoring solvent stabilization (solvation), the stability of ions is a function of charge density. A small ion has a higher charge density than a larger ion of the same charge, making the smaller ion less stable. From the covalent radius of oxygen compared with sulfur, and fluorine compared with chlorine, it can be estimated that the charge density on the larger atom is half that of the smaller. The resulting stabilization of the conjugate base more than compensates for the decrease in electronegativity in moving down the column; so H2S is a stronger acid than H2O, and HCl a stronger acid than HF. Since sulfur and chlorine are nearly the same size (covalent radii being 1.02 ±0.02 Å), electronegativity explains the difference in acidity between H2S and HCl.
If the heavy atom of an acid carries a formal charge, its acidity will be changed substantially. This is demonstrated by the examples on the right. Ammonium and hydronium ions carry a positive charge, and the acidity of the species is increased by over fifteen powers of ten relative to uncharged ammonia and water. By contrast, hydrogen sulfide and hydrogen selenide are dibasic acids (they have two acidic protons). Once the first proton has been lost, the acidity of the negatively charged conjugate base is reduced over a million fold. This is true for most other dibasic acids such as H2SO4 and H2CO3.
|
NH4(+)
9.24 |
OH3(+)
-1.74 |
S-H(–)
15 (pK2) |
Se-H(–)
11 (pK2) |
Accurate acidity measurements in the pKa range from 1 to 14 can usually be made in water solution. However, acids stronger than the hydronium ion (H3O(+)) and bases stronger than hydroxide ion (OH(–)) react immediately with this solvent, and the resulting "leveling effect" prevents direct measurement of their pKa's. One way of circumventing this difficulty is to examine the acidity of very strong (pKa < 0) and very weak (pKa > 15) acids in different (non-aqueous) solvents, and to extrapolate these measurements to water. For example, solvents such as acetic acid, acetonitrile and nitromethane are often used for studying very strong acids. Very weakly acidic solvents such as DMSO, acetonitrile, toluene, amines and ammonia are used to study the acidities of very weak acids. The errors introduced in extreme cases, such as methane, are often large; but the overall range of acid strengths observed in this manner cannot be questioned.
Hybridization
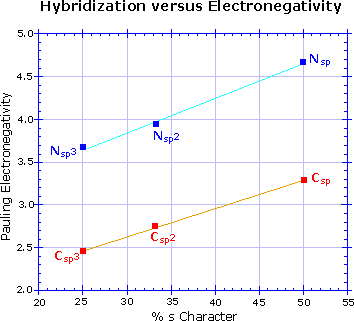
Hybridization has a strong influence on acidity, as shown by the three carbon acids on the upper
left below. The greater the s-character of the orbital holding the electron pair of the conjugate
base, the greater will be the stability of the base. This corresponds to the lower energy of an
s-orbital compared with p-orbitals in the same valence shell. It also corresponds to the increased
electronegativity or inductive electron withdrawal that is found for different hybridization
states of a given atom, as depicted in the graph on the right. The difference in acidity of
2-butynoic acid and butanoic acid, shown in the shaded box at lower left, provides a further
illustration of this inductive effect.
Carbocation stability is also influenced by hybridization, but in the
opposite direction (sp3 > sp2 > sp).
Carbon Acids
Inductive Effect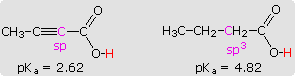
|
|
|||||||||
Stereoelectronic Control of Enolization
Many carbon acids have enhanced acidity because of a neighboring functional group. The acidity of
alpha hydrogens in
aldehydes, ketones and
esters is well
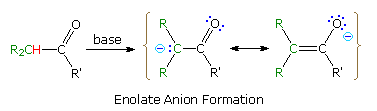
documented, and is the source of many important synthetic procedures. The following equation
illustrates the general enolate anion transformation, with the acidic alpha-hydrogen colored red.
The resulting ambident anion is
stabilized by charge delocalization, and may react with electrophiles at both carbon and
oxygen.
Stereoelectronic factors govern the enolization reaction, as illustrated by clicking on the
diagram below. The bond from the alpha carbon to the acidic alpha-hydrogen must be oriented 90° to
the plane of the carbonyl group, or parallel to the pi-electron system (colored magenta here). The
ideal overlap occurs with a 0° dihedral angle between this bond and the pi-orbital, as shown.
By clicking on the diagram a second time, the importance of this stereoelectronic requirement will be demonstrated. An increase in the acidity of carbon acids activated by two carbonyl groups is well known, and is illustrated by the two beta-dicarbonyl compounds on the left side of the diagram. In such cases the acidic C-H unit may be oriented perpendicular to both carbonyl groups, and the resulting planar anion is stabilized by additional charge delocalization (over both oxygens and the central carbon). In the case of the bicyclic diketone on the right, the C-H bond nearly eclipses the two carbonyl C-O bonds, resulting in a dihedral angle with the pi-electron systems of roughly 90°. Consequently, the acidity of this hydrogen is similar to that of the hydrogens of an alkane or cycloalkane. It should also be apparent that if an enolate anion were to be formed to the bridgehead carbon, the double bond would be prohibited by Bredt's rule.
The trends outlined here are a bit oversimplified, since solvent and cation influences have been ignored. For a discussion of these factors, and practical applications of enolate anion intermediates in synthesis Click Here
A more extensive discussion on acidity is available one the Acid-Base Page.