Frontier Molecular Orbitals
A useful molecular orbital model for analyzing pericyclic reactions has been proposed by Kenichi Fukui of Japan. This frontier-orbital approach is based on the assumption that bonds are formed by a flow of electrons from the highest occupied molecular orbital (HOMO) of one reactant or participating bond to the lowest unoccupied molecular orbital (LUMO) of another reactant or bond. To illustrate, consider the [4+2] cycloaddition of 1,3-butadiene and ethylene to give cyclohexene. The pertinent molecular orbitals involved in this reaction were described elsewhere, and the two combinations of HOMO and LUMO are shown in the following diagram. Note that regardless of which combination is examined, the terminal orbital phases match, indicating a bonding interaction. Since the dienophile often has electron-withdrawing substituents and the diene is usually electron rich, the electron flow pattern on the left seems to best represent the course of most Diel-Alder reactions.
![Frontier-orbital [4s+2s] Diels-Alder: two HOMO-LUMO combinations of butadiene and ethylene with matching terminal phases](/reusch/virtualtext/img/supfacy1.gif)
This frontier orbital approach to cycloaddition reactions is general, and is simple to apply
thanks to the alternation of terminal orbital phase relationships as a polyene changes from a 4n
electron system to a 4n + 2 electron system. By clicking on the above diagram, these phase
relationships will be displayed for HOMO and LUMO of polyenes in both classes. Only the terminal
orbital phases (colored in the diagram) are important for frontier orbital analysis. The frontier
orbital analysis of a [6s + 2s] cycloaddition reaction will be demonstrated
by clicking on the diagram a second time. An antibonding node is present in both HOMO-LUMO
combinations (one is shown), so this reaction is orbital symmetry forbidden.
An additional
feature of this treatment of cycloaddition reactions is its rationalization of the tendency of
Diels-Alder reactions of cyclic dienes to form
endo adducts preferentially. This
was noted earlier, and is further
illustrated in the following diagram. The first two equations are straightforward examples of the
endo predilection of substituents or rings (colored green) attached to a bicyclic ring system. The
third equation shows a more subtile case of the same orientational factor, which essentially
favors that [4+2] transition state in which unsaturated substituents on the dienophile are
directed toward the diene double bonds. By clicking on this diagram, the secondary orbital bonding
interaction that stabilizes the endo transition state for a typical Diels-Alder reaction will be
displayed.
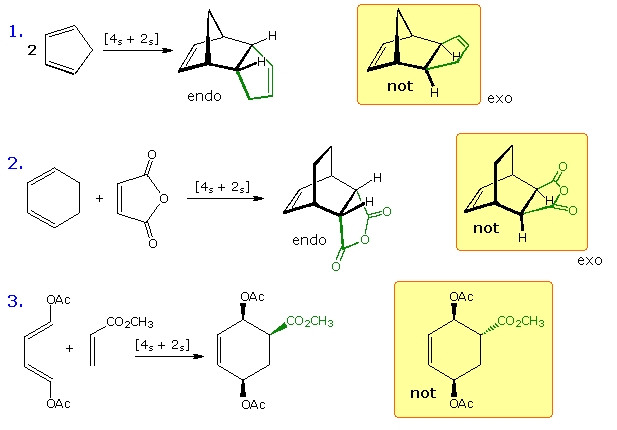
Not all cycloaddition reactions favor endo products. The predominant product from the [6s + 4s] reaction shown earlier is the exo adduct. Frontier orbital analysis of this case demonstrates that secondary orbital interaction destabilizes the endo transition state.
Electrocyclic Reactions
The stereochemistry of electrocyclic reactions is easily predicted by frontier orbital analysis. Two examples are shown in the following diagram. The upper reaction represents the thermal interconversion of 1,3-butadiene and cyclobutene; the lower reaction shows the similar interconversion of 1,3,5-hexatriene and 1,3-cyclohexadiene. The HOMO orbital of the open chain isomer for each example is displayed on the left. In order to close the ring, the terminal p-orbital components of this orbital must be rotated so that identical phased lobes can interact to form a new sigma-bond (green line). It should be evident that the orbitals of the upper example must rotate in the same direction (conrotatory), either clockwise or counter-clockwise, to permit this bonding to occur. The terminal orbitals of the lower example must rotate in opposite directions (a disrotatory motion) to achieve the same bonding interaction. The alternation of terminal orbital phases in the HOMO of 4n and 4n+2 polyenes, as noted above, is therefore a predictor of the general course of electrocyclic reactions.
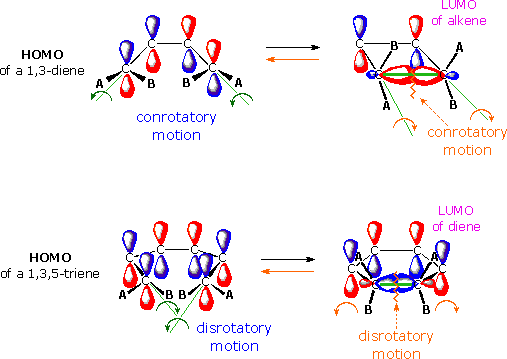
The reverse ring opening electrocyclic process (orange arrows) is conveniently treated by assuming
a flow of electrons from the HOMO of the sigma bond to the LUMO of the π-electron system. Of
course, the same configurational motion is predicted by this analysis, and is in fact required by
the principle of microscopic reversibility.
To examine a model of the p-orbital components of 1,3,5-hexatriene pi-orbitals.
Click Here
To examine the actual molecular orbitals of 1,3,5-hexatriene Click Here
Sigmatropic Reactions
Accounting for the facility of [1,5] hydrogen shifts in contrast to the rarity of documented [1,3]
shifts is a sine qua non of pericyclic reaction theory. One frontier orbital approach to
these reactions establishes the sigma C–H bond as the HOMO site, and the adjacent pi-orbital(s) as
the LUMO. In the following diagram these entities are defined for both the general [1,5] and [1,3]
relationships. Since it is necessary for the origin and terminus of a hydrogen shift to be near
each other, a potential [1,5] system must be coiled in an appropriate manner (top-central formula)
for such a rearrangement to occur. As shown, there is a phase correlation of HOMO and LUMO
termini, rendering these reactions symmetry allowed. Note the stereoelectronic requirement
that the sigma bonds be oriented parallel to the pi-orbital system. The lower row shows a similar
analysis of the suprafacial [1,3] shift, which is found to be symmetry forbidden. A
previously described [1,7]
hydrogen shift is antarafacial with respect to the triene moiety, and is therefore symmetry
allowed.
If the π-electron system is
electronically excited by
the absorption of light, the LUMO becomes the next higher energy orbital, and [1,3] shifts are
symmetry enabled. By clicking on the diagram an interesting example of such a rearrangement will
be displayed. Sigmatropic [1,5] hydrogen shifts are prohibited in this example, because the diene
is constrained in a s-trans-configuration so that origin and terminus of such a shift are kept far
apart. The conjugated diene chromophore absorbs UV-light, and once the [1,3] shift has occurred
the unconjugated double bonds no longer absorb 245 nm light. Two hydrogens at C-6 are candidates
for this shift, but only the axial hydrogen (colored orange) has the necessary parallel
orientation (a stereoelectronic discrimination). Other photochemical products were formed in this
reaction, and these may include an isomeric diene formed by a [1,3] shift of the axial C-12
hydrogen.
![Suprafacial sigmatropic H shifts: allowed [1,5] (matched phases) vs forbidden [1,3] (node) frontier-orbital analysis](/reusch/virtualtext/img/fo-sigm1.gif)
When considering the sigmatropic shift of an alkyl group, such as methyl, the possibility of antarafacial bonding to carbon must be considered. Although such rearrangements are rare, they have been observed along with the expected inversion of configuration at the migrating group. By clicking on the above diagram a second time, the general form of these rearrangements will be shown together with two examples. The [1,3] shift of a methyl group (colored green) is pictured at the top. The phases of the central p-orbital component are colored light blue and orange, rather than blue and red, to avoid confusion with the pink and purple phases of the methyl carbon p-orbital in the transition state. Clearly, symmetry allowed 1,3-bonding takes place with inversion at the methyl carbon. In the two example shown at the bottom (A & B) the migrating group is marked by an asterisk.
The most commonly used sigmatropic reactions are those involving a [3,3] shift. Of these, the Cope
rearrangement of 1,5-dienes is a prominent example, and the orbital assignments for this reaction
are shown in the following diagram. One of the terminal double bonds (for our purpose it doesn't
matter which) is defined as the LUMO partner. The central sigma bond (joining C-3 and C-4 of the
diene), together with the remaining double bond, is then the HOMO for this analysis. As shown on
the top of the diagram, the [3,3] shift is found to be symmetry allowed.
An alternative
interpretation is shown in the shaded box. Here, the 1,5-diene is dissected into two allylic
radicals. Because an allylic radical has three π-electrons, the HOMO is π2. The central
carbon atom of this fragment is the locus of a node, so the terminal carbons have opposite phases.
Bonding at both ends of the bis-allylic intermediate is therefore allowed.
![Frontier-orbital analysis of the allowed [3,3] sigmatropic shift, with the bis-allyl radical interpretation](/reusch/virtualtext/img/fo-33sg1.gif)
The spatial orientation of the 1,5-diene may assume either a chair-like or boat-like transition
state configuration. These possibilities will be displayed by clicking on the diagram. In each
case the HOMO and LUMO components are identified, and the orbital lobes in the chair drawing are
shaded to show their relative orientation. Specific cases proceeding by both transition states are
known, but in general, acyclic reactants prefer the chair-like pathway. The boat-like transition
state is possibly destabilized by a non-bonding secondary interaction involving orbitals at C-2
and C-5.
Thermal rearrangement of the diastereomeric 3,4-dimethyl-1,5-hexadienes to isomeric
2,6-octadienes clearly shows a preference for a chair-like transition state. These reactions will
be displayed above by clicking on the diagram a second time. The top row illustrates reaction
paths for the racemic diastereomer (R = CH3). The conformational equilibrium between
the diaxial conformation shown left of center and the diequatorial conformer to its right will
strongly favor the latter (>99%). Assuming similar activation energies for [3,3] sigmatropic
shifts from each, the formation of (E,E)-2,6-octadiene is expected to predominate. The meso-isomer
depicted on the left of the second row exists as a mixture of equivalent axial-equatorial
conformers, each of which rearranges to (E,Z)-2,6-octadiene. Rearrangement of these diastereomers
by way of a boat-like transition state would generate a different set of products, as shown on the
left of the third row for the meso isomer. The data in the following table clearly show a strong
preference for a chair-like transition state, when that path is available to a rearranging system.
|
Cope Rearrangement of racemic and meso-3,4-Dimethyl-1,5-Hexadiene to 2,6-Octadiene |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
Finally, the example on the right of the second row demonstrates that a [3,3] sigmatropic rearrangement may serve to transmit chirality from an existing stereogenic center to one that is newly formed. Once again, chair and boat-like transition states control this transfer in a different manner.
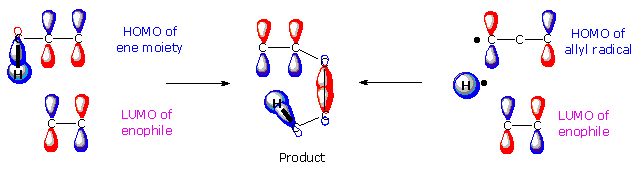
Ene Reactions
Since ene reactions are often stereospecific, and do not seem to proceed by way of discrete intermediates, they are sometimes grouped together with other pericyclic reactions. A frontier orbital analysis of the forward ene reaction is shown in the following diagram., and displays many features of the [3,3] sigmatropic shift. Thus, hydrogen atom transfer from an allylic site to a double bond is seen to be symmetry allowed with respect to the HOMO of an allyl radical and the LUMO of an alkene (right side of diagram). There is also symmetry correlation between the HOMO and LUMO terminal sites of the ene and enophile reactants, as defined on the left. Ene reactions proceed best when the enophilic double bond is electron deficient, and the ene reaction is often catalyzed by Lewis Acids.