Vinylagous Systems
A vinylagous relationship is one in which a double bond extends by conjugation an interaction between two sites in a molecule, or between two reacting species. There are many examples of this phenomenon, as the following discussion will demonstrate.
Hydroxyl Acidity
The carboxyl group is an outstanding example of the interaction of two functional groups (hydroxyl
and carbonyl) when they are bonded together. One manifestation of this interaction is the enhanced
acidity of the carboxyl group relative to an isolated hydroxyl group (more than ten powers of
ten). This increase in acidity was explained earlier by
resonance stabilization of the carboxylate anion
conjugate base. As shown below (first equation), this results in negative charge delocalization
over two oxygens as compared with full charge localization on an alkoxide oxygen.
A
carbon-carbon double bond can conjugatively link hydroxyl and carbonyl groups so that the
corresponding alkoxide base is similarly stabilized by charge delocalization. The second equation
illustrates this vinylagous relationship, and a green box identifies the double bond that
establishes the vinylagous link. A third resonance contributor, which has the negative charge on
the central carbon atom, has been omitted from this drawing, but is an important factor in
alkylation reactions of beta-dicarbonyl compounds.
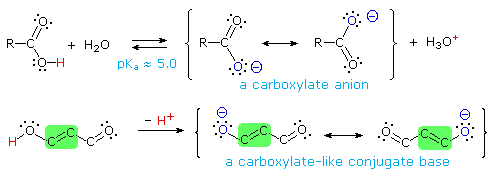
Some examples of vinylagous acids are shown below, together with their pKa values. The first compound, the enol of 1,3-cyclohexanedione, fits the general example outlined above, and has an acidity comparable to acetic acid. The second example (tropolone) is triply vinylagous. Although it is less acidic than a carboxylic acid, it is much more acidic than an alcohol and even a thousand times greater than phenol.
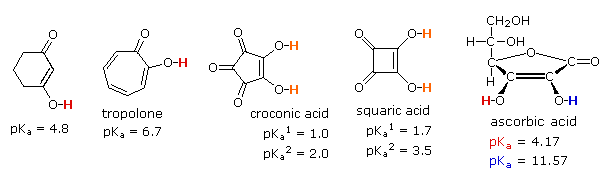
The third and fourth examples listed are extraordinary cases in which vinylagous activation enhances the acidity of two hydroxyl functions in the same molecule. Because of symmetry, it is not possible to identify which is more acidic, but the first pKa ranks these acids stronger than phosphoric acid. The high acidity of the second hydroxyl function is even more surprising, and undoubtedly reflects the fact that both conjugate bases are stabilized by charge delocalization over a different set of oxygen atoms. The case of ascorbic acid, commonly known as Vitamin C provides the last example. Here it should be clear that the beta-hydroxyl group (the red hydrogen) is vinylagously activated by the carbonyl function, and is a stronger acid than acetic acid. The alpha-hydroxyl group (blue hydrogen) is part of an enol, and is therefore expected to have an acidity similar to phenol.
If the hydroxyl group of a vinylagous acid is replaced by an amino group or an alkoxyl group the resulting compounds may be classified as vinylagous amides and esters respectively. The following general formulas illustrate these terms. As expected from the electron interactions shown by the resonance formulas, the properties of such compounds are similar to amides and esters. The basicity of a vinylagous amide, for example, is much less than that of an enamine.
|
Vinylagous Amide |
R2N–C=C–C=O |
|
R2N(+)=C–C=C–O(–) |
|---|---|---|---|
|
Vinylagous Ester |
RO–C=C–C=O |
|
RO(+)=C–C=C–O(–) |
β-Elimination Reactions of Carbonyl Derivatives
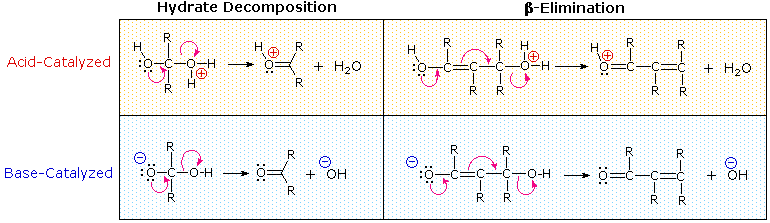
The facile elimination of water from aldol products was noted earlier. Either acid or base catalysis is effective, suggesting that enol species may be involved in these elimination reactions. As shown in the following diagram, these enolic intermediates are vinylogues of carbonyl hydrates, a function known to undergo rapid and reversible loss of water.
Conjugate Addition Reactions
One of the largest and most diverse classes of reactions is composed of nucleophilic additions to a carbonyl group. Both reversible and irreversible addition reactions have been described, and in all cases the initial step involved covalent bonding of a nucleophile to the electrophilic carbon atom of the carbonyl group. As noted earlier, conjugation of a double bond to a carbonyl group transmits the electrophilic character of the carbonyl carbon to the beta-carbon of the double bond. A resonance description of this transmission is shown below. From this formula it should be clear that nucleophiles may bond either at the carbonyl carbon, as for any aldehyde, ketone or carboxylic acid derivative, or at the beta-carbon. These two modes of reaction are referred to as 1,2-addition and 1,4-addition respectively, and will be displayed here when the "Nucleophilic Addition" button is clicked.
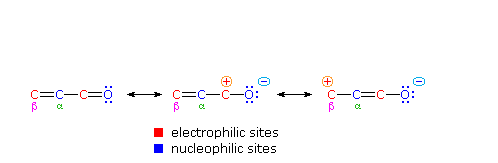
The nucleophile in this scheme is shown with a negative charge, which is neutralized in the addition products by treatment with water. Neutral nucleophiles such as 1° and 2°-amines may also add in the same manner, and do not require a neutralization step. The term "1,4-addition" is applied to the product of conjugate addition (initial nucleophile bonding at the beta-carbon) because the product initially formed is presumably the unstable enol tautomer.
Reversible addition reactions of nitrogen, oxygen and sulfur nucleophiles to unsaturated carbonyl and nitrile compounds normally give 1,4-addition products rather than their 1,2-addition isomers. This preference for conjugate addition may be attributed in part to the thermodynamic advantage of addition reactions to carbon-carbon double bonds over additions to a carbonyl function. This factor was noted earlier in the chapter on aldehydes and ketones. Although nucleophilic addition reactions to alkenes are usually slow, conjugation with a carbonyl or nitrile function vinylagously activates the beta-carbon, resulting in rapid addition. It is likely that rapid 1,2-addition occurs as well, but because it is reversible, the thermodynamically favored 1,4-product accumulates. Several examples of these conjugative addition reactions are given below.
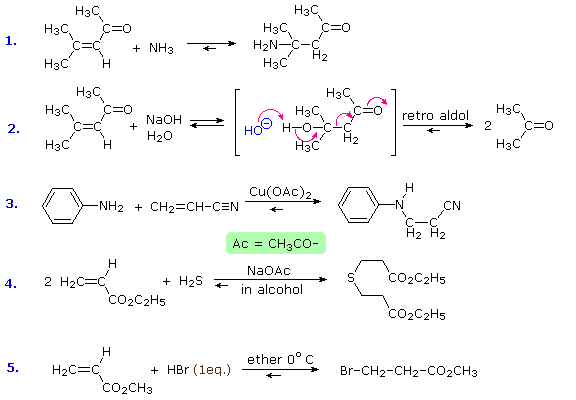
The reaction of 4-methyl-3-penten-2-one with hydroxide ion (# 2) is interesting because the 1,4-addition product is the aldol product from acetone. A retro (or reverse) aldol reaction generates acetone as the chief product. The third and fourth reactions demonstrate the use of acetate salts as catalysts for some conjugative additions, and the last reaction is an acid-catalyzed 1,4-addition (bromide anion is the nucleophile).
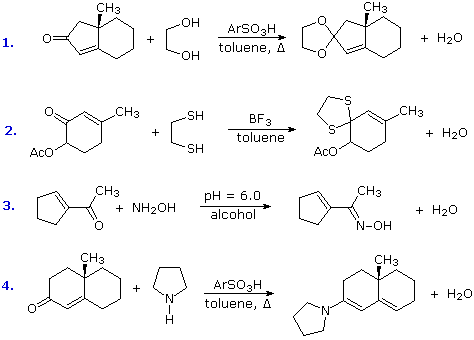
Some typical aldehyde and ketone substitution reactions that proceed from 1,2-addition intermediates still take place in the expected manner when conjugated double bonds are present. Most of these involve a final dehydration that is only possible if an initial 1,2-addition has occurred. As demonstrated by the following examples, acetals (# 1 & 2), imine derivatives (# 3) and enamines (# 4) can all be prepared in the usual way.
The Michael Reaction
Conjugative addition of carbon nucleophiles to unsaturated esters, ketones, nitriles, sulfones and other activated double bonds is a useful synthetic method known as the Michael reaction. In combination with alkylations and condensations, the Michael reaction may be used to construct a wide variety of complex molecules from relatively simple starting materials. The carbon nucleophiles used in the following examples include cyanide ion, sodium diethylmalonate and the conjugate base of cyclohexane-1,3-dione. These anions are sufficiently stable that their addition reactions may be presumed reversible. If this is so, the thermodynamic argument used for hetero-nucleophile additions would apply here as well, and would indicate preferential formation of 1,4-addition products. Cyanide addition does not always follow this rule, and aldehydes often give 1,2-products (cyanohydrins). In each case the initial reaction is a Michael addition, and the new carbon-carbon bond is colored magenta. Any subsequent bonds that are formed by other reactions are colored orange.
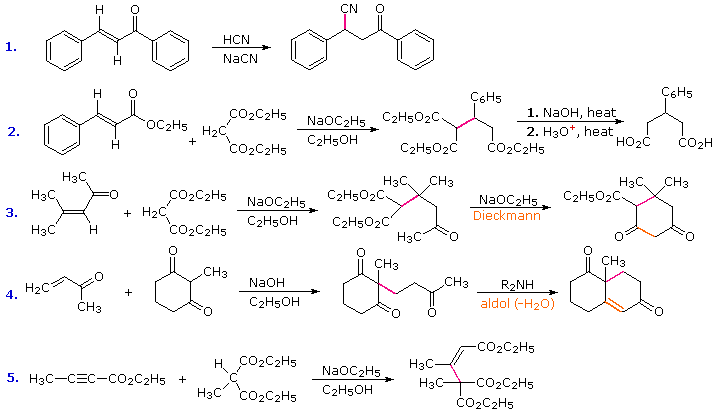
In all the above examples the vinylagous electrophile (Michael acceptor) is drawn on the left, and the carbon nucleophile (Michael donor) is to its right. By clicking the "More Examples" button, four additional Michael reactions will be displayed. These illustrate the use of unsaturated nitrile and extended vinylagous acceptors, and enamine and nitroalkane donors.
Irreversible Addition Reactions
Some reagents, such as metal hydrides and organometallic reagents, add to aldehydes, ketones and
esters in an irreversible fashion,
and it is likely that similar reactions of vinylogous functions will also be irreversible. Since
1,2-additions to the carbonyl group are fast, we would expect to find a predominance of
1,2-products from these reactions.
For the hydride reductions shown in the first three
equations below, this is the case. However, not all compounds of this kind give clean
1,2-reduction. Lithium aluminum hydride often reacts further with allylic alcohols, reducing the
carbon-carbon double bond as well. It must therefore be used with care. Sodium borohydride may
also give conjugate addition products in some cases. Fortunately, this can be prevented by adding
cerium trichloride (CeCl3) to the reaction mixture. If the 1,4-reduction product is
desired it is best obtained by using a
dissolving metal reduction.
The remaining five equations displayed here describe the use of various organometallic reagents. Alkyl lithium compounds usually give 1,2-addition products, as shown in equation # 4. Grignard reagents, on the other hand, may add in both a 1,2- and 1,4-manner, depending on the substitution at the electrophilic sites. Unsaturated aldehydes usually give 1,2-addition, as in equation # 5. An equivalent ketone having a large carbonyl substituent, as in equation # 6, gives 1,4-addition, and if the isopropyl group is replaced by a smaller methyl group a nearly 50:50 mixture of 1,2- and 1,4-addition products is obtained. Grignard reactions may be shifted to a 1,4-addition mode by adding copper salts, but a better strategy is to use a Gilman reagent, as shown in the last two equations. The metal enolate that results from this conjugate addition may be quenched by hydrolysis, as in equation # 7, trapped as a silyl enol ether, as in equation # 8, or alkylated by a suitable alkyl halide.
Vinylagous Activation of C-H
The increased acidity and reactivity of C-H bonds alpha to a carbonyl group has been described. This characteristic is critical to useful synthetic reactions such as the aldol and Claisen condensations, as well as enolate alkylation. A vinylagous or doubly vinylagous relationship results in a similar activation of a more remote carbon, which is illustrated by the resonance structures in the green shaded box below. Here the negative charge on the conjugate base is delocalized at three atoms, the oxygen and the alpha and gamma carbons. Some reactions of such extended enolate anions with electrophilic reagents are shown below the resonance diagram.
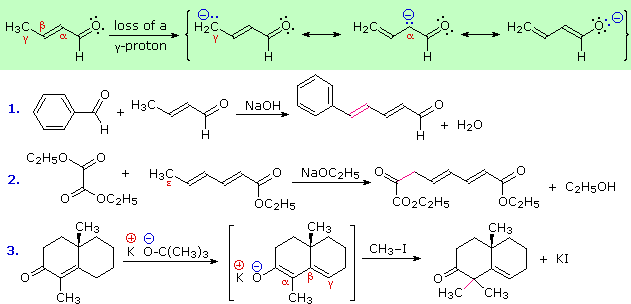
Examples # 1 and 2 correspond to aldol and Claisen condensations respectively. The new carbon bonds are colored magenta. In the first case the dienolate anion can react with an electrophile at either the alpha or gamma carbons. The reversibility of the aldol reaction favors formation of the most stable product, which is the extended conjugated dienal formed by condensation at the gamma location. The second condensation is also reversible and takes place at the end (epsilon) carbon. The third example is an alkylation and is irreversible. Reaction is fastest at the alpha carbon atom of the dienolate anion, and once an alkyl group is bonded there it will not change location.
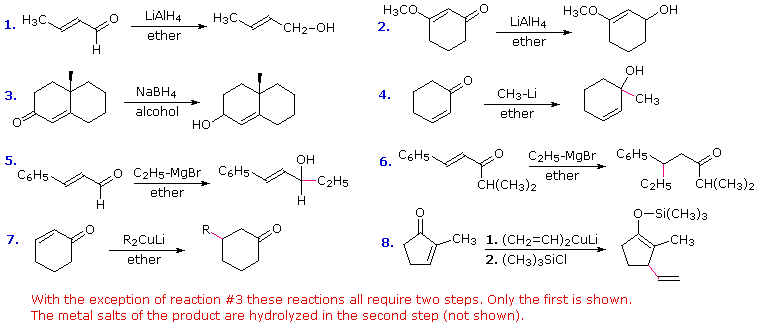
Practice Problems
Eight reactions using chemical reactions similar to those described above and in the dissolving metals reduction section are displayed here. Answers will be given by clicking the appropriate button.