Ozonolysis
Ozone, an allotrope of oxygen, is a 1,3-dipole that undergoes [4s + 2s] cycloaddition to alkenes. The structure of ozone may be written as a resonance hybrid of zwitterionic structures, as shown here. Although the energetically favored structures on the left are 1,2-dipoles, ozone can only react as a terminal 1,3-dipole, since the central oxygen has a filled valence shell.
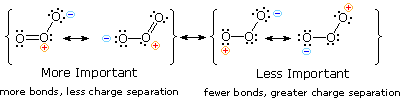
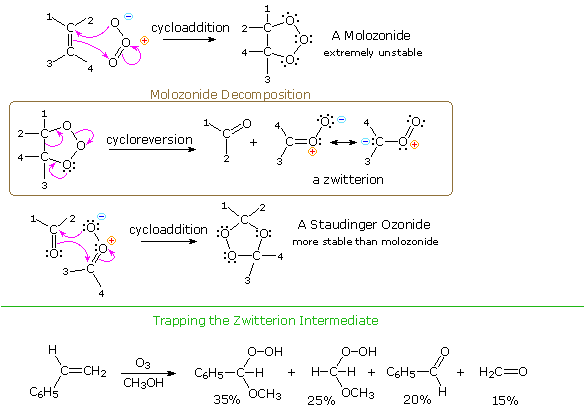
The initial product of ozone cycloaddition to an alkene is called a molozonide. Molozonides
are very unstable and rapidly decompose; nevertheless, spectroscopic evidence for the transient
existence of a molozonide has been obtained at -100 °C. The manner in which molozonides decomposes
has been the subject of many investigations. As shown in the following diagram, it is usually
depicted (the Criegee mechanism) as a concerted cycloreversion leading to a carbonyl fragment and
a zwitterionic species, sometimes referred to as a carbonyl oxide. Calculations indicate an
activation energy of 20 kcal/mol or less for such a transformation. An alternative cleavage of one
of the two very weak O–O bonds in the molozonide, followed by immediate fragmentation of the
resulting diradical would lead to equivalent intermediates. The zwitterionic intermediate is a
1,3-dipole of the same type as ozone; however, it prefers to cycloadd to carbonyl functions rather
than alkene double bonds. In this way an isomeric ozonide, known as a Staudinger ozonide, is
formed. Such ozonides are much more stable than their molozonide precursors, and in some cases may
be isolated as pure crystalline compounds. In general, ozonides must be handled with care, since
they may decompose explosively.
If ozonolysis is carried out in an alcohol solvent, instead
of the customary methylene chloride solvent, the dipolar carbonyl oxide may be trapped as the
α-hydroperoxide of an ether. The reaction of styrene, shown below the green line, is an example.
In the case of unsymmetrical alkenes such as styrene, the carbonyl oxide zwitterion is stabilized
by alkyl substituents on the positively charged carbon, and the fragmentation of the molozonide
favors this intermediate. Likewise, nearby electron withdrawing substituents destabilize the
carbonyl oxide species.
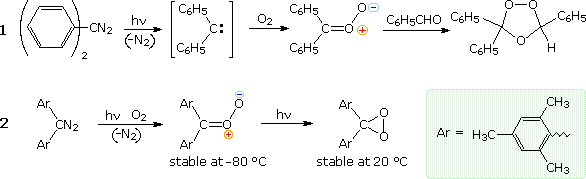
In addition to the alcohol trapping results, there is a growing body of evidence supporting key
elements of the mechanism shown here. Some examples will be displayed above by
clicking on the mechanism diagram. Thus, the carbonyl oxide zwitterion undergoes
cycloaddition reactions with extraneous carbonyl compounds, as shown by ozonide C in equation 1.
Although two zwittterions could combine to give a dimeric bis-peroxide, the extreme reactivity of
these intermediates makes such an encounter improbable, compared with other reactive interactions.
Dialkyl substitution serves to improve the stability of the carbonyl oxide, and in the case of
reaction 1, the bis-peroxide B is obtained in small amounts together with the ozonides A and C.
Intramolecular
recombination of the carbonyl and carbonyl oxide fragments is usually favorable, as the ozonolysis
of cyclopentene (equation 2) demonstrates. The bicyclic [3.2.1] ozonide is obtained pure in yields
up to 80%. Curiously, cyclohexene does not yield a similar monomeric ozonide, but instead, an
assortment of oligomeric ozonides and peroxides. An explanation for this behavior may reside in
the configuration of the intermediate carbonyl oxide zwitterion. Just as
oximes and other imine derivatives
may exist in syn and anti stereoisomeric configurations, the RHC=O–O grouping may
adopt similar structures. The resonance description of a carbonyl oxide is presented in the gray
shaded box above. Of the four Lewis structures shown, the most favorable is clearly the one on the
left. The C=O–O unit is planar and bent, with a bond angle ca. 120 °. If the barrier to rotation
(or inversion) about the C=O bond is sufficiently high, the carbonyl oxide will have a distinct
configuration relative to the substituents on carbon. The example shown here is syn. If a
syn configuration of the carbonyl oxide is required for intramolecular cycloaddition of
short chains, and the cyclohexene molozonide fragments to an anti isomer, its failure to
form a monomeric ozonide is understandable. A beautiful demonstration supporting this explanation
will be displayed above by clicking on the mechanism diagram a second time. Cyclopentene
and cyclohexene derivatives, each carrying an appropriately sized, deuterium labeled, aldehyde
substituent, were ozonized in methylene chloride at -78 °.C. Molozonide fragmentation in each case
produces a carbonyl oxide having two equal length aldehyde chain substituents. Unlike the
unsubstituted examples noted above, both compounds lead to monomeric ozonides in good yield. If
the carbonyl oxide intermediate is formed in a stereo-random fashion, or if the isomeric forms are
rapidly interconverted, then the deuterium label will be scrambled between the bridgehead location
and the remaining aldehyde side chain. The data presented in the diagram clearly demonstrates
stereoselective fragmentation of each molozonide and a strong preference for
syn-cycloaddition.
Equation 1 in the following diagram illustrates formation of a typical carbonyl oxide intermediate by oxygen addition to a carbene, generated by photochemical elimination of nitrogen from a diazo compound. This carbonyl oxide exhibits the same reactivity as those formed by ozonolysis, including cycloaddition to an aldehyde carbonyl function. In the same manner, a sufficiently stable carbonyl oxide species, permitting spectroscopic characterization, was prepared recently, as shown in equation 2. Although still highly reactive, this intermediate could be examined in solution at temperatures below -80 °C. Further irradiation isomerized the zwitterion to its neutral dioxetane isomer, shown on the right. Despite the apparent ring strain of this compound, it is stable up to 20 °C and could be crystallized. Spectroscopic and X-ray diffraction data confirm the structure shown here.
In most synthetic applications of ozonolysis, oxidative or reductive decomposition of the Staudinger ozonide to carbonyl products or their acetal derivatives is the final stage of the reaction. Two common methods employed in such work-up were described in an earlier section of this text, and many others have proven useful. For example, quenching the ozonolysis reaction mixture in a THF solution of lithium aluminum hydride results in reduction of both carbonyl moieties to alcohols. A particularly useful set of conditions that permit two symmetrically equivalent aldehyde functions to be released in different forms or oxidation states is shown in the following diagram. These procedures all begin with a low temperature ozonolysis in the presence of methanol. Once the double bond is completely converted to the initial ozonide product, as evidenced by the characteristic blue color of unreacted ozone, the excess ozone is removed by a stream of nitrogen. At this point one of the aldehydes is free and the other exists in the form of an α-hydroperoxide methyl ether. In procedure A, addition of p-toluenesulfonic acid converts the free aldehyde to a dimethyl acetal. The acid catalyst is then neutralized with sodium bicarbonate, and the hydroperoxide is reduced to a hemiacetal by treatment with dimethyl sulfide, a generally useful reductant for ozonides or peroxides. This reduction is shown by the upper equation in the blue shaded box.
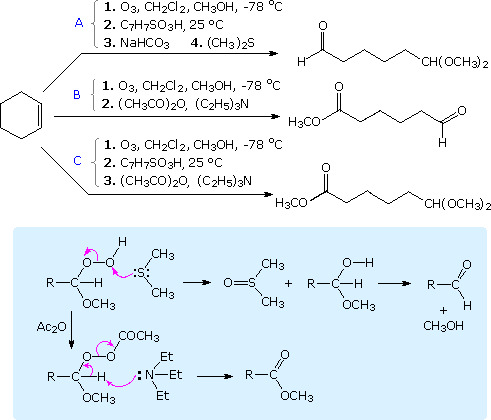
Following the initial ozonolysis, procedures B and C proceed by first removing excess methanol as a benzene azeotrope. The key reaction in both cases is an eliminative oxidation of the α-hydroperoxide methyl ether, as shown by the bottom reaction in the shaded box. This reaction is effected either immediately (conditions B) or following acetal formation as in procedure A (conditions C).