Photochemistry
The study of chemical reactions, isomerizations and physical behavior that may occur under the influence of visible and/or ultraviolet light is called Photochemistry. Two fundamental principles are the foundation for understanding photochemical transformations:
The efficiency with which a given photochemical process occurs is given by its Quantum Yield (Φ). Since many photochemical reactions are complex, and may compete with unproductive energy loss, the quantum yield is usually specified for a particular event. Thus, we may define quantum yield as "the number of moles of a stated reactant disappearing, or the number of moles of a stated product produced, per einstein of monochromatic light absorbed.", where an einstein is one mole of photons. For example, irradiation of acetone with 313 nm light (3130 Å) gives a complex mixture of products, as shown in the following diagram. The quantum yield of these products is less than 0.2, indicating there are radiative (fluorescence & phosphorescence) and non-radiative return pathways (green arrow). The primary photochemical reaction is the homolytic cleavage of a carbon-carbon bond shown in the top equation. Here the asterisk represents an electronic excited state, the nature of which will be defined later.
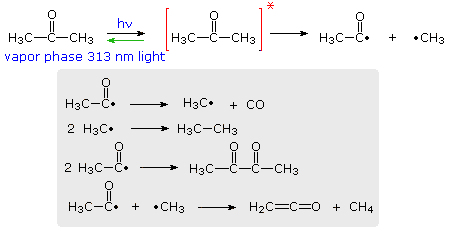
Several secondary radical reactions then follow (shown in the gray box), making it difficult to assign a quantum yield to the primary reaction. The biacetyl product, formed in the third reaction, may itself be excited by light or accept excitation energy from an excited acetone molecule, further complicating this process.
By comparison, the light induced chlorination of methane, or other alkanes, has a large quantum yield, often near 106, because of the secondary chain reactions that follow the primary cleavage of the Cl-Cl bond. Equations illustrating such halogenation reactions have been presented elsewhere in this text.
Mechanistic Background
| Type of Glass | Wavelength Cut-off |
|---|---|
| Pyrex | < 275 nm |
| Corex | < 260 nm |
| Vycor | < 220 nm |
| Quartz | < 170 nm |
The light required for a photochemical reaction may come from many sources. Giacomo Ciamician, regarded as the "father of organic photochemistry", used sunlight for much of his research at the University of Bologna in the early 1900's. Depending on the compounds being studied and the information being sought, bright incandescent lamps (chiefly infrared and visible light), low, medium and high pressure mercury lamps (185 - 255 nm, 255 -1000 nm & 220 -1400 nm respectively), high intensity flash sources and lasers have all been used. In careful studies of specific chromophores, sources of monochromatic light may be desired.
In this section we shall focus chiefly on the nature and behavior of the electronic excited states formed when a photon is absorbed by a chromophoric functional group. As a rule, such excitation results in a change in molecular orbital occupancy, an increase in energy, and changes in local bonding and charge distribution. The principles set forth in the UV-Visible Spectroscopy chapter will provide a helpful foundation.
We begin by considering the electronic excitation of a simple diatomic molecule such as Cl2
or Br2. Both absorb light, chlorine in the 300 to 380 nm region. and bromine in the 360
to 510 nm region.
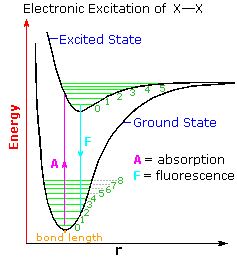 The diagram on the right illustrates the initial electronic excitation. Both the ground (lowest
energy electronic state) and excited states are shown as energy profiles populated by vibrational
energy states (green lines) as well as rotational states (not shown). The electron reorganization
that occurs when the ground electronic state is excited by absorption of a photon takes place much
more rapidly than any movement of the atom nuclei that eventually follow. In other words, electron
shifts, when viewed from the perspective of the nuclear coordinates, occur as if the heavier
nuclei were fixed in place. This consequence of the Born-Oppenheimer approximation led James
Franck and R. Condon to formulate the Franck-Condon Principle:Electronic transitions occur much faster than nuclei can respond.
The diagram on the right illustrates the initial electronic excitation. Both the ground (lowest
energy electronic state) and excited states are shown as energy profiles populated by vibrational
energy states (green lines) as well as rotational states (not shown). The electron reorganization
that occurs when the ground electronic state is excited by absorption of a photon takes place much
more rapidly than any movement of the atom nuclei that eventually follow. In other words, electron
shifts, when viewed from the perspective of the nuclear coordinates, occur as if the heavier
nuclei were fixed in place. This consequence of the Born-Oppenheimer approximation led James
Franck and R. Condon to formulate the Franck-Condon Principle:Electronic transitions occur much faster than nuclei can respond.
Overall bonding in an excited state is usually lower than in the ground state. Thus, the X–X
bond length is increased in the excited state. At normal temperatures essentially all molecules
will exist in the ground vibrational state (zero level). The Franck-Condon principle requires that
excitation occur by a vertical transition, shown by the red line, resulting in the population of
higher vibrational levels in the excited state. Several events may then take place.
2. The excited state may return to the ground state by emitting a photon (light blue line). If this happens from the zero vibrational level the frequency or energy of the emitted light will be lower than that of the initially absorbed light. This radiative decay is called fluorescence if it takes place rapidly from the initial excited state. It is termed phosphorescence if it occurs slowly by way of other excited states.
3. If a higher vibrational level of the excited state is populated, either by the initial Franck-Condon transition or by collisional activation, the molecule may cleave into two X• atoms. Note that vibrational level 5 of the excited state is roughly coincident with bond breaking.
Virtually all organic compounds have more than two atoms, so the potential energy state diagram of
X2 must be adjusted for the increased number of bonding relationships. One way of doing
this is to retain the energy coordinate while dispensing with the dimensional (r) coordinate. The
Jablonski diagram shown below is an example, in which the spatial orientation of the
various electronic states is not specified. Nevertheless, Franck-Condon transitions are
expected.
One important feature conveyed by the diagram is that more than one electronic
excited state is likely to exist for a given molecule, six are drawn and labeled in the diagram.
Each electronic state will have a group of vibrational (and rotational) states, depicted by light
blue lines above each state marker. Transitions between electronic states often occur to higher
vibrational levels which then relax to lower levels by collisional loss of heat (translational
energy).
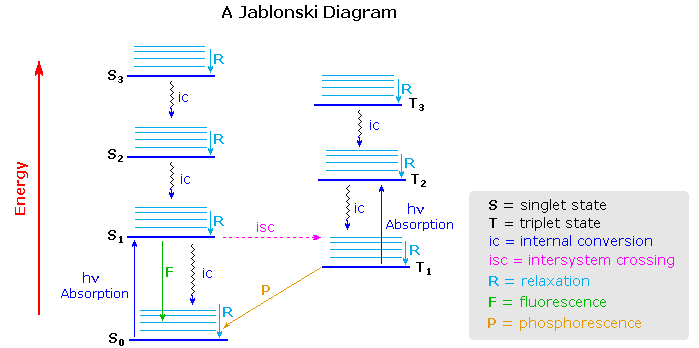
Excited states may be classified as singlet or triplet based upon their electron
spin angular momentum. The electrons in most non-metallic organic compounds are paired (opposite
spins) in bonding and non-bonding orbitals, resulting in a net zero spin diamagnetic molecule for
the ground state. Such states have a single energy state in an applied magnetic field, and are
called singlets. Electronic states in which two electrons with identical spin occupy different
orbitals (the Pauli exclusion principle) have a net spin of 1 (2 • 1/2) and are paramagnetic. In a
magnetic field such states have three energy levels (+1, 0, -1) and are called triplets. Molecular
oxygen
is a rare example of a triplet ground electronic state.
The distinction between singlet and triplet states is important because photon
induced excitation always leads to a state of the same multiplicity, i.e. singlet to singlet or
triplet to triplet. Since most ground states are singlets, this means that the excited states
initially formed by absorption of light must also be singlets. Internal conversion of
excited states to lower energy states of the same multiplicity takes place rapidly with loss of
heat energy (relaxation). Alternatively, an excited state may return to the ground state by
emitting a photon (radiative decay). In the study of acetone described above,
nearly 80% of the excited singlet states lose energy by internal conversion and about 3% by
fluorescence. Conversion of a singlet state to a lower energy triplet state, or vice versa, is
termed intersystem crossing and is slower than internal conversion. Radiative decay from a
triplet state is called phosphorescence and is generally quite slow. The approximate timescales
for these transitions are given in the following table.
| Process | Transition | Timescale (sec) |
|---|---|---|
| Light Absorption (Excitation) | S0 → Sn | ca. 10-15 (instantaneous) |
| Internal Conversion | Sn → S1 | 10-14 to 10-11 |
| Vibrational Relaxation | Sn* → Sn | 10-12 to 10-10 |
| Intersystem Crossing | S1 → T1 | 10-11 to 10-6 |
| Fluorescence | S1 → S0 | 10-9 to 10-6 |
| Phosphorescence | T1 → S0 | 10-3 to 100 |
| Non-Radiative Decay |
S1 → S0 T1 → S0 |
10-7 to 10-5 10-3 to 100 |
The non-radiative decay noted in the last row may take place by intermolecular energy transfer to a different molecule. This collisional process is termed quenching if the focus is on the initially excited species, or sensitization if the newly created excited state is of interest. Photochemical sensitization commonly occurs by a T1 + S0 → S0 + T1 reaction, where the bold red-colored species is the sensitizer. The new triplet excited state may then undergo characteristic reactions of its own.
Photochemical Reactions
Alkene Isomerization
A photochemical reaction occurs when internal conversion and relaxation of an excited state leads
to a ground state isomer of the initial substrate molecule, or when an excited state undergoes an
intermolecular addition to another reactant molecule in the ground state. The cis-trans
photochemical isomerization of stilbene is a reaction of the first kind, as shown in the following
diagram. Both cis and trans-stilbene undergo π → π* electron excitation by absorption of uv light.
Whereas isolated double bonds require 180 nm light for such excitation, conjugation with the
phenyl substituents
lowers the transition energy
to about 300 nm, a more easily achieved source. The
molar absorptivity of the
cis-isomer is less than that of the trans-isomer because steric crowding of the ortho sites causes
the phenyl groups to twist slightly out of coplanarity.
The stability of the stereoisomers of stilbene is due to a 62 kcal/mole
barrier to rotation about the double bond produced by the π-bond. This bonding is absent in the π
→ π* excited state (magenta curve in the diagram). Both the initial S1 states formed
from the cis and trans ground states are slightly twisted (the cis by 25° & the trans by 13°)
with the C=C double bond being lengthened by about 4.5%. These local S1 states quickly
relax to a common lower energy twisted configuration (θ ≅ 90°). Non-radiative internal conversion
of this S1 twisted state leads to the transition state region of S0, which
decays equally to the ground states of the cis and trans isomers. This simple configurational
isomerization about a single double bond is referred to as a One-Bond-Flip (OBF) event.
A
small proportion (6%) of the trans-S1 state fluoresces back to the trans-isomer, but
there is less than 0.1% fluorescence from the cis-S1
state. Triplet states (not shown) may also be formed, but there is no observable phosphorescence
in solution, and non-radiative decay from such states results in similar cis-trans isomerization.
The quantum yield of trans to cis isomerization decreases in viscous solvents, accompanied by an
increase in fluorescent decay. Freezing the solutions to a rigid glass halts the isomerization and
results in maximum fluorescence efficiency.
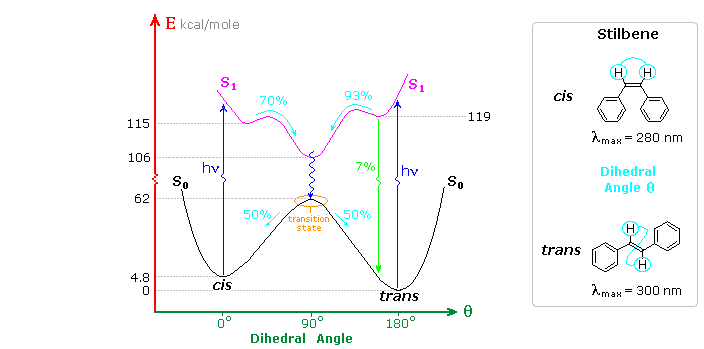
Inspection of the cis-S1 local minimum near θ = 0° shows that only 70% of the molecules
in this state relax to the lower energy twisted S1 state. The remaining 30% undergo an
electrocyclic rearrangement to an
isomeric 4a,4b-dihydrophenanthrene (DHP) S1 state, as illustrated above by clicking on
the diagram. Molecules occupying this new excited state then relax to either DHP or cis-stilbene
ground states. Over time, DHP accumulates and may be converted to phenanthrene by mild oxidation
with air or iodine. Substituted phenanthrenes thus become available from trans-stilbene precursors
prepared by Wittig or Grignard procedures.
Another minor product from the photolysis of
stilbene has been identified as 1,2,3,4-tetraphenylcyclobutane (distilbene), shown in the
following diagram. Unlike the previously described unimolecular isomerizations, this compound is
formed by a bimolecular reaction between an electronically excited stilbene and a ground state
stilbene molecule. The dimer accumulates slowly, irradiation of a 0.07 M solution of
trans-stilbene in benzene producing 27% (as two stereoisomers) after two months of exposure. As
expected, higher stilbene concentrations increase the rate of dimer formation. Thus, a 0.56 M
solution of trans-stilbene in ethyl acetate yielded 11% dimer in four hours of irradiation.
Mercury lamps were used as the uv light source in all these cases.
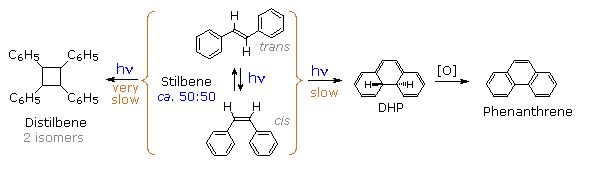
Electron donating substituents on the benzene rings facilitate the dimerization, as does restricting the ability of the double bond to isomerize or achieve orthogonally twisted excited states. Examples of these influences will be displayed above by clicking on the diagram.
The stilbene reactions described above have been attributed to singlet excited states. Triplet excited states exist, but their formation by intersystem crossing is inefficient. In order to study the behavior of triplet excited states it is often necessary to generate them by energy transfer from a higher triplet excited state of a suitable sensitizer molecule. Spin conservation requires that spin exchange take place during the collisional energy transfer. Thus, a sensitizer triplet (Zt), generated from sensitizer molecule Zs, reacts with a ground state stilbene molecule (Ms) in the following manner:
The following diagram illustrates important features of this sensitization reaction. The ideal sensitizer (Z) absorbs light preferentially with respect to the substrate (M), and undergoes efficient intersystem crossing to a triplet excited state (T1). This energetic state then serves to activate a substrate molecule to a lower energy triplet state by collisional exothermic energy and spin exchange, returning the sensitizer to its ground state. A variety of useful sensitizers have been identified, and by clicking on the diagram a few of these will be drawn below.
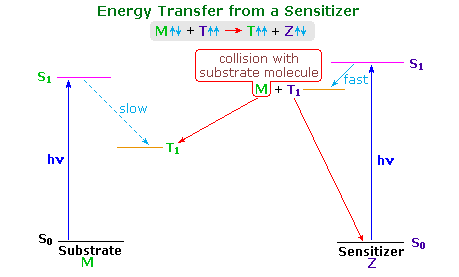
In the early 1960's Prof. George Hammond's group at California Institute of Technology undertook an extensive study of the photosensitized isomerization of stilbene and related alkenes and dienes.
| Sensitizer | Triplet Energy | Cis:Trans Ratio |
|---|---|---|
| benzophenone | 69 kcal/mole | 60:40 |
| benzil | 53 kcal/mole | 85:15 |
| pyrene | 47 kcal/mole | 90:10 |
| eosin | 43 kcal/mole | 0:100 |
Carbonyl Compounds
Background
Conjugated ketones are often good sensitizers, thanks to the efficiency of intersystem crossing
from the n → π* singlet to the triplet. However, before discussing this application it will
be helpful to consider the bonding structure of the carbonyl group itself. In the case of the
simple compound formaldehyde, the Lewis formula consists of two C–H sigma bonds, a C–O sigma bond,
a C=O pi bond and two non-bonding electron pairs.
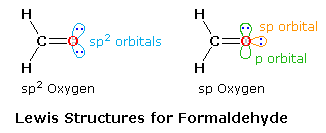 As shown in the diagram on the right, two Lewis structures, differing in the hybridization of
oxygen, may be drawn, The structure on the left is a common representation in which an sp2
oxygen replaces one of the CH2 groups of an ethylene molecule. However, based on the
principle that stable molecules will adopt the strongest bonding possible, the right hand
structure becomes an attractive alternative. An sp2–sp sigma bond should be stronger
and shorter than a sp2–sp2 sigma bond, and the shorter bond distance will
enhance the pi-bonding. Double bonds are of course shorter and stronger than equivalent single
bonds. The average values given in the following table indicate that C=C is 13% shorter and 76%
stronger than a C–C bond, whereas C=O is 15% shorter and over 100% stronger than a C–O bond,
possibly reflecting the sp hybridization of the oxygen. An important difference between these
models is found in the nature of the non-bonding electron pairs on oxygen. In the
sp2-oxygen model these occupy very similar (degenerate) orbitals, but in the sp-oxygen
model one pair is in a relatively low energy sp-orbital and the other in a higher energy
p-orbital. Molecular orbital calculations clearly show that the latter model is a better
representation than the former.
As shown in the diagram on the right, two Lewis structures, differing in the hybridization of
oxygen, may be drawn, The structure on the left is a common representation in which an sp2
oxygen replaces one of the CH2 groups of an ethylene molecule. However, based on the
principle that stable molecules will adopt the strongest bonding possible, the right hand
structure becomes an attractive alternative. An sp2–sp sigma bond should be stronger
and shorter than a sp2–sp2 sigma bond, and the shorter bond distance will
enhance the pi-bonding. Double bonds are of course shorter and stronger than equivalent single
bonds. The average values given in the following table indicate that C=C is 13% shorter and 76%
stronger than a C–C bond, whereas C=O is 15% shorter and over 100% stronger than a C–O bond,
possibly reflecting the sp hybridization of the oxygen. An important difference between these
models is found in the nature of the non-bonding electron pairs on oxygen. In the
sp2-oxygen model these occupy very similar (degenerate) orbitals, but in the sp-oxygen
model one pair is in a relatively low energy sp-orbital and the other in a higher energy
p-orbital. Molecular orbital calculations clearly show that the latter model is a better
representation than the former.
| Bond Length, Å | C–C, 1.54 | C=C, 1.34 | C–O, 1.44 | C=O, 1.22 |
| Bond Energy, kcal/mole | C–C, 84 | C=C, 146 | C–O, 86 | C=O, 177 |
To examine the molecular orbitals of formaldehyde Click Here.
A general diagram illustrating the two major electronic excitations of a carbonyl group is shown below. This terminology has been defined in the UV-Visible Spectroscopy chapter. The weak nature of the n → π* absorption is due to the poor overlap of the p-orbital housing the higher energy electron pair with the π* orbital (they are essentially orthogonal). In contrast, π and π* orbitals overlap almost completely, so the probability of a π → π* excitation being achieved by a photon of proper energy is high. This spatial overlap requirement has been termed a selection rule by spectroscopists. A second important selection rule concerns the probability of electronic transitions between states of different spin multiplicity, the spin selection rule. This rule reflects the high probability of transitions between different singlet states, or between different triplet states, but the low probability of singlet-triplet or triplet-singlet transitions. A table summarizing these rules is given below.
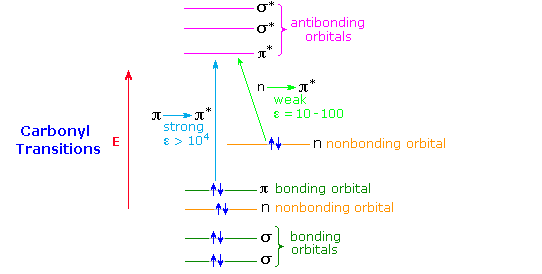
Formaldehyde, acetaldehyde and acetone show strong π → π* absorption at 180 to 190nm (ε = 103 to 104), and weak n → π* absorption at 280 to 300 nm (ε = 12 to 20). Conjugation with the π-electrons of a double bond or a benzene ring shifts both absorptions to longer wavelength and increases the strength of absorption.
| Compound | Cyclohexanone | (CH3)2C=CHCOCH3 | C6H5CHO | C6H5COCH3 |
|---|---|---|---|---|
| λmax π → π* | 200 nm (ε 2000) | 230 nm (ε 12,300) | 242 nm (ε 14,000) | 238 nm (ε 13,000) |
| λmax n→ π* | 285 nm (ε 14) | 325 nm (ε 90) | 328 nm (ε 50) | 320 nm (ε 40) |
Aryl ketones such as acetophenone (right hand compound in the table above), undergo rapid
intersystem crossing of the n → π* singlet excited state to an energetically close π
→ π* triplet state. The latter then quickly decays to the lower energy n → π*
triplet, as shown for benzophenone in the diagram below. This circuitous route for the low
probability direct conversion reflects modified selection rules formulated by Prof. M. A. El Sayed
of UCLA (last two entries in the following table). This pathway is not available to most aliphatic
ketones, so their intersystem crossing rates from n → π* singlets to triplets are slow.
As
noted in the diagram, there is very little fluorescent decay from the S1 state, and
radiationless decay to S0 is less than 1% of the intersystem crossing to T2.
In the absence of quenching reactions, T1 returns to S0 by a mixture of
phosphorescent and radiationless decay.
|
Benzophenone Excited States
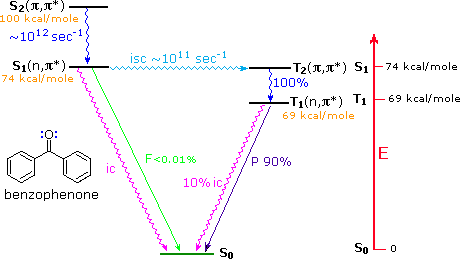
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The n → π* excited states of carbonyl compounds display a rich chemistry in their own
right.
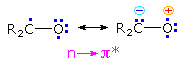 Since the oxygen has an unpaired electron, it behaves in much the same way as an alkoxy radical,
as noted in the chapter on
free radical chemistry. Hydrogen abstraction
and addition to double bonds are typical reactions. Cleavage of neighboring carbon-carbon bonds
may also occur, the two most common of these being designated Type I and Type II.
General equations for these primary reactions, starting from aryl ketone excited states, are
described in the following diagram. The n → π* excited state shown in brackets at the
center of the diagram may also be described as the resonance hybrid drawn on the right. Note that
the ground state polarity of the carbonyl group has been reversed in this excited state.
Since the oxygen has an unpaired electron, it behaves in much the same way as an alkoxy radical,
as noted in the chapter on
free radical chemistry. Hydrogen abstraction
and addition to double bonds are typical reactions. Cleavage of neighboring carbon-carbon bonds
may also occur, the two most common of these being designated Type I and Type II.
General equations for these primary reactions, starting from aryl ketone excited states, are
described in the following diagram. The n → π* excited state shown in brackets at the
center of the diagram may also be described as the resonance hybrid drawn on the right. Note that
the ground state polarity of the carbonyl group has been reversed in this excited state.
The
triplet excited state of aryl ketones has a lifetime of about 100 ns, and most of the subsequent
reactions are very fast (rates range from 104 to 109 M-1
sec-1). Consequently, the effect of quenchers such as 1,3-pentadiene on product
distribution provides valuable information about the mechanisms. These quenchers have T1
energies much lower and S1 energies higher than corresponding ketone states. The rate
of quenching is nearly diffusion controlled, and is therefore proportional to the concentration of
the quencher. Since the ground state triplet of oxygen reacts rapidly with triplet excited states,
air must be excluded when studying these reactions.
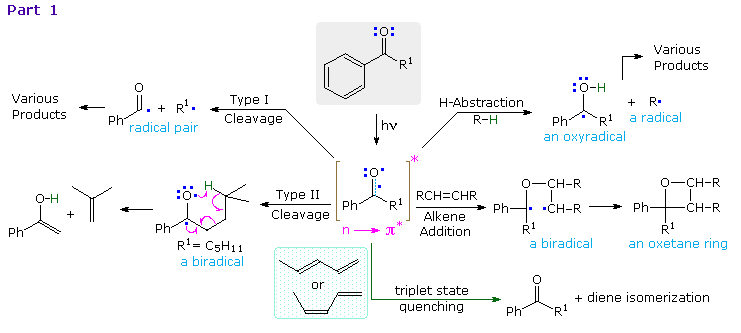
Examples of hydrogen abstraction and alkene addition reactions will be displayed above by clicking
on the diagram. Reduction of benzophenone to benzpinacol is common in most hydrocarbon solvents,
even in pure benzene. The diphenylmethanol radical formed by hydrogen abstraction is relatively
stable and, once formed, dimerizes to benzopinacol in nearly quantitative yield. In contrast, the
more reactive isopropanol radical is converted to acetone by transferring its hydroxyl hydrogen to
benzophenone, thus forming another diphenylmethanol radical.
The photoaddition of carbonyl
compounds to olefins had been observed by the Italian chemist E. Paterno in 1909, but the
structure of the oxetane products remained unknown until elucidated by Büchi and coworkers
(M.I.T.) in 1954. This reaction is now known as the Paterno-Büchi reaction. The mechanism
shown here was established for the electron-rich alkene dioxene, a compound that favors electron
transfer to benzophenone. Other alkenes are also believed to react by way of a 1,4-biradical
intermediate, and significant regioselectivity is found with many unsymmetrical alkenes. Two
additional examples of this reaction are drawn in the gray-shaded box at the bottom. The second
illustrates the intramolecular variant. A related example of carbonyl addition to alkenes is the
use of benzophenone as a light activated initiator of polymerization for monomers such as styrene
and methyl methacrylate.
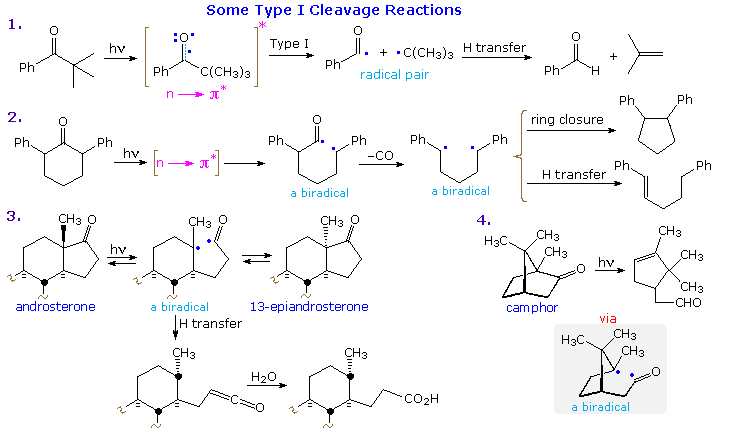
Examples of type I fragmentations are displayed below. These reactions are fast, and often proceed
with a high quantum yield. The first example, shown at the top, illustrates a typical homolytic
cleavage of a carbonyl substituent, followed by a fast hydrogen atom transfer between the caged
radical pair. Three intramolecular type I reactions, all proceeding by way of biradical
intermediates, demonstrate the variety of products that may be formed by hydrogen transfer
disproportionation and/or ring closure. The cyclic transition states for such intramolecular
hydrogen transfer usually include five or six, and occasionally seven atoms.
By clicking on
the diagram, examples of type II reactions will be displayed. These reactions all begin by an
intramolecular hydrogen atom abstraction, similar to the intermolecular abstraction in the
reductive dimerization of benzophenone.The general example at the top of the
diagram shows how a key 1,4-biradical intermediate, formed by intramolecular hydrogen abstraction,
may disproportionate to several different products, including type II cleavage to the enol
tautomer of a ketone (green shaded box) and ring closure to a cyclobutanol (the
Yang Reaction). In hydrocarbon solvents intramolecular hydrogen transfer back to carbon is
common, reducing the quantum yield of these products by over 50%. This reverse hydrogen transfer
is inhibited by hydrogen bonding solvents, which stabilize the newly formed OH group. As expected,
the rate at which the biradical is formed is sensitive to substitution at the γ-carbon. If R=H (a
methyl group) the rate is 100 times slower than if R=CH3 (abstraction of a 3°-
hydrogen). This reactivity order reflects the bond dissociation energies of the C–H bonds in the
same fashion as noted for
free radical halogenation of alkanes.
Also, replacement of all γ-hydrogens by deuterium reduces the rate of biradical formation
substantially.
The ratio of type II cleavage and Yang cyclization products from a given substrate depends on several factors. The steroid ketone in example 2 gives both reactions, with cyclization predominating. Example 3 is instructive, first because the rate of biradical formation is much higher than in many other cases, as a result of conformational bias favoring the reactive orientation (as drawn); and second, because cyclobutanol formation induces severe strain. Only type II fragmentation is observed. Finally, example 4 proceeds by a δ-hydrogen abstraction (7-membered cycle), as the only possible transformation of its kind. The resulting 1,5-biradical then closes to the product shown. Remarkably, phosphorescence studies indicate that this reaction occurs from a low-lying π→ π* triplet state, rather than the n→ π* triplet usually associated with this class of reaction.
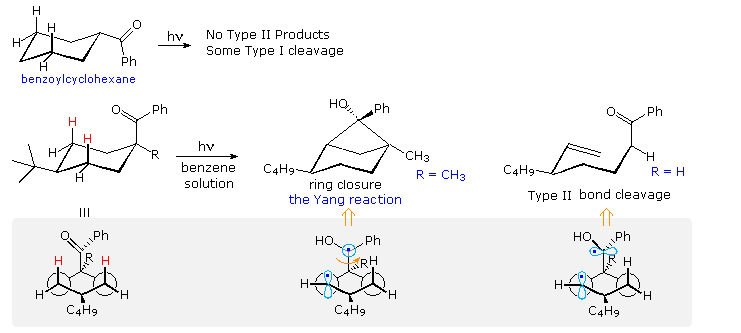
Type II reactions depend, of course, on close approach of the carbonyl oxygen to a γ-hydrogen. The conformational mobility of the examples drawn above permits this to occur within the lifetime of the excited triplet. However, it is not difficult to find molecules for which this is not possible. Benzoylcyclohexane, shown at the top of the following diagram, is such a compound. Because of its equatorial location on the six-membered ring, the carbonyl oxygen cannot reach any of the γ-hydrogens. If the benzoyl group is forced into an axial orientation by a large cis-tert-butyl group on c-4, then the oxygen can easily reach the axial cis γ-hydrogens (colored red. The resulting type II photochemistry depends on the other C-1 substituent. If R = H, type II cleavage is the only product; however this changes to exclusive stereoselective cyclization if R = CH3. The conformational drawings in the gray-shaded box suggest a reason for this remarkable change. Following the initial hydrogen abstraction, the 1,4-biradical is well aligned for ring closure, but the benzyl radical must be rotated 90° before it is properly aligned for bond cleavage (far right structure). A C-1 methyl substituent hinders this rotation, leading to formation of the strained cyclobutanol product.
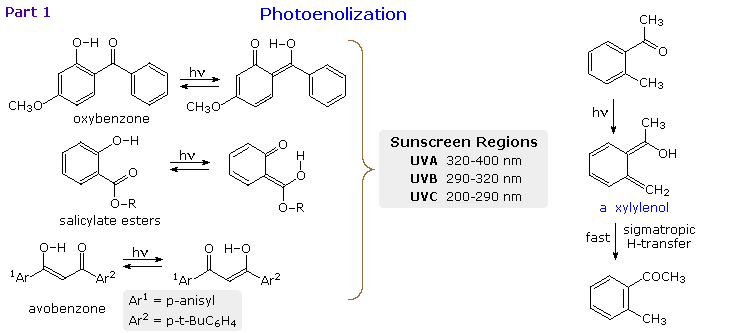
When the reactive carbonyl function and a γ-hydrogen are conjugated via an aromatic ring or double
bond, the 1,4-diradical created by hydrogen abstraction quickly relaxes to a conjugated enol
tautomer. This is illustrated in the following diagram for a β-hydroxyl substituent, examples on
the left, and an ortho-tolyl ketone on the right. If an aromatic ring has been disrupted by the
photoenolization, as in all the cases but avobenzone (lower left), the enoltautomer is unstable
and rapidly reverts to the initial aromatic carbonyl compound. This might appear to be a useless
transformation, but it finds practical application as a sunscreen ingredient. The three compounds
on the left are examples of current sunscreen components. Oxybenzone screens UVA, salicylate
esters (R=C8 to C10) screen UVB and avobenzone screens both.
Careful
studies of the photoenolization of ortho-alkyl benzophenones and acetophenones have enhanced our
understanding of this deceptively simple transformation. By clicking on the diagram the behavior
of ortho-methylacetophenone will be elaborated. Excitation of the anti and syn mixture of this
ketone leads to two stereoisomeric n→ π* triplet states, TE and TZ
respectively. The former has a much longer lifetime (τ) than the latter since it must undergo a
conformational change before γ-H abstraction from the ortho-methyl substituent can take place. In
both instances a twisted triplet biradical is formed. This biradical may be intercepted by
electron transfer to PQ2+ followed by rapid proton loss. This event is easily monitored
by the intense blue color of the PQ+• radical cation, and serves to set the lifetime of
the biradical at 580 nsec. Relaxation of the biradical leads to a mixture of (E) and
(Z)-xylylenols, the latter undergoing rapid H-transfer back to the starting ketone. (Z)-Xylylenol
formation directly from a syn n→
π* singlet state is possible and would not be detected.
Depending on the reaction conditions, (E)-xylylenols may undergo a variety of reactions or slowly
relax back to the initial ketone. Clicking on the above diagram a
second time will show examples of other reactions (Part 3). Unlike
benzophenone itself, the ortho substituted compound in the upper left corner does not undergo any
pinacol reduction. Instead, γ-H abstraction produces a mixture of
interconverting twisted biradicals which decay predominantly to (E,E) and
(Z,E)-xylylenols. Only the former is sufficiently long lived to yield distinctive products.
At temperatures below 0 °C
conrotatory electrocyclization
forms cis-1,2-diphenylbenzocyclobutenol in high yield. At room temperature or above, conrotatory
ring opening regenerates the (E,E)-xylylenol which may be trapped by cycloaddition to
maleic anhydride.
Competition between photoenolization and type II cleavage has been explored
in the manner shown above, by clicking on the diagram a
third time (Part 4). The syn conformer of butyl (and isoamyl)
o-tolyl ketone gives exclusive photoenolization, 60% from the n → π* singlet state, the
corresponding triplet state undergoing xylylenol formation faster than type II cleavage. The anti
conformer reacts exclusively from the triplet state, which either isomerizes to the syn triplet or
generates the biradical precursor to type II products. As noted in the previous section, t-butanol
favors type II cleavage. The 8-methyl-1-tetralone served as a reference molecule, constrained with
the carbonyl oxygen syn to the methyl substituent.
Conjugated π-Orbital Functions
Dienes and Polyenes
Acyclic conjugated dienes, trienes and polyenes absorb strongly in the 220 to 300 nm region of the
near ultraviolet. The resultant singlet excited states undergo a variety of reactions, as shown in
the following diagram for 1,3,5-hexatriene and two 2,5-dialkyl derivatives. Two configurational
isomers (E & Z) are possible, together with three coplanar s-cis and s-trans
conformations for each (the
cEt structure is not drawn). The E-configuration is normally
more stable than its Z-isomer, and for R=H or CH3 the elongated
tEt conformation is preferred. The various conformers of both the
E & Z-isomers are rapidly interconverted at room temperature, the equilibrium
composition depending on the size of substituents such as R. The light induced isomerization about
the central double bond of the trienes (e.g. E→Z) is an example of
OBF.
Now, electronic π → π* excitation of any triene increases
the π-bonding between carbons 2 & 3 as well as 4 & 5, as may be seen in the
HOMO (π3) and LUMO (π4*) molecular orbitals
of the parent triene. Taken together with the very short lifetimes of these excited states (≤10
nsec), this suggests that photochemical products should reflect the rotamer composition of the
ground state. Put another way, excited state rotamers are not expected to equilibrate prior to
reaction, the NEER principle (Non-Equilibration of Excited Rotamers). An example of NEER is
found in the absence of 1,3-cyclohexadiene among the photoproducts from the parent triene (R=H),
compared with its increased quantum yield as R is changed to methyl and then tert-butyl
(increased cZc concentration).
For E as well as
Z-trienes, reactivity parallels an increase in the population of non-planar s-cis rotamers.
Furthermore, 1,2-divinylcyclopentene (drawn in the gray-shaded box) is photochemically unreactive
and exhibits fluorescence in solution, demonstrating that twisting about the central (3,4-) double
bond of the triene is an essential factor in these reactions.
Irradiation of trans,cis,cis-1,3,5-cyclononatriene, shown at the bottom
of the diagram, by 254nm light establishes a 60:40 photoequilibrium with the cis-fused
bicyclononadiene on its right. This equilibrium is shifted to the right by longer wavelength 300nm
light, which also induces further electrocyclization to tricyclo[4.3.0.0]non-3-ene. As a rule,
cyclohexadienes absorb light at longer wavelength than similar non-cyclic hexatrienes. Higher
temperatures permit the thermal electrocyclization of trans,cis,cis-1,3,5-cyclononatriene
to the trans-fused bicyclononadiene on its left.
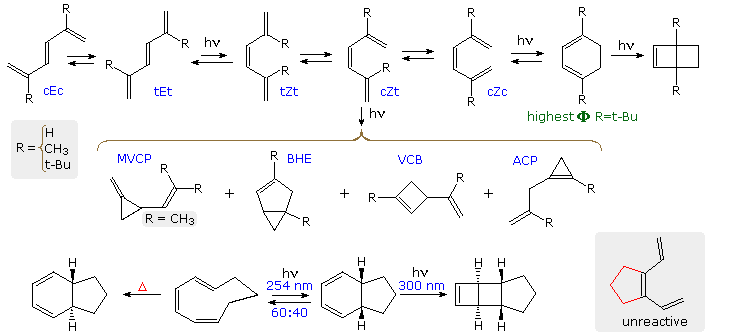
Finally, diradical mechanisms for the formation of allylcyclopropene (ACP), vinylcyclobutene (VCB) and bicyclohexene (BHE) products will be shown above by clicking on the diagram. ACP may be formed from either the tEt or cZt excited states, but VCB and BHE require the latter rotamer. The MVCP product on the left is unique to R = CH3.
As noted above, the reduced bond order of a formal double bond in its first
excited state leads to the OBF mechanism for isomerization in fluid solution. The torsional
movement in this change is sensitive to the viscosity of the solution and to other structural
constraints that may exist. Thus, the facile isomerization and weak fluorescence, typical for
trans-stilbene in fluid solutions, change to low isomerization and strong fluorescence in rigid
media at low temperatures. Here, the rapid radiationless deactivation of the excited state by OBF
is impeded and a normally non-fluorescent compound becomes fluorescent.
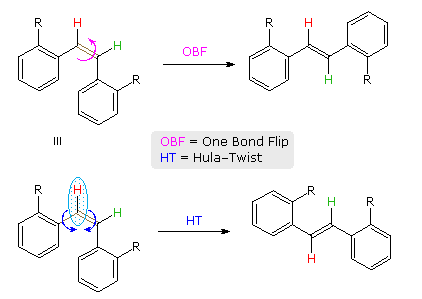
A more complex,
higher energy motion that achieves double bond isomerization in restrictive environments has been
described and named the Hula-Twist. As shown for a cis-stilbene derivative in the following
diagram, the OBF process involves a 180° flip of the aryl group on the right side of the double
bond. By contrast, the HT process proceeds by simultaneous rotation of a small C–H moiety (light
blue ellipse) about the double bond and an adjacent single bond (both colored brown). This not
only converts the cis-isomer to its trans-configuration, but also effects a
conformational change (note the orientation of the red hydrogen relative to the aryl substituent
on its left).
The difference between OBF and HT isomerization of a Z to E-triene will be shown above by clicking on the diagram. It has been proposed that photoisomerization of biologically important polyenes, such as retinal and calciferol may occur by the HT mechanism because translocation of a single H-atom is less volume demanding than the turning over of one-half the molecule, as required by the OBF process. This would be especially important when the chromophore is encapsulated in a protein or bilayer membrane.
Instructive examples of the photochemical behavior of biologically important polyenes may be examined by clicking these buttons.
Enone Reactions
[2+2]-CycloadditionIn the earlier discussion of stilbene photoisomerization a [2+2]-cyclodimerization was noted. The construction of cyclobutane rings from alkene components is rare in ground state chemistry, so a general photochemical synthesis of this kind would provide a valuable addition to our assortment of useful carbon-carbon bond forming reactions. Double bonds activated by conjugation with a carbonyl group have proven especially effective in this respect, both in forming [2+2] dimers and adding to isolated alkene functions. A particularly important example of photodimerization involves the damage to DNA caused by uv light. Thymine (or uracil in the case of RNA) is one of four heterocyclic base components of nucleic acids. On exposure to ultraviolet light, thymine may undergo [2+2]-dimerization with a suitably oriented nearby thymine, as shown in the diagram below. When this happens toxic products are generated that are directly responsible for cell death via mutagenic action, suppression of DNA transformation and/or activation of carcinogenic pathways. A natural repair mechanism employing a photolyase enzyme system reverses the dimerization.
![UV-induced [2+2] dimerization of two thymine bases to a cyclobutane-linked cis-syn thymine dimer](/reusch/virtualtext/img/thydim1.gif)
Four possible dimers of thymine (and uracil) may be formed. The cis-syn isomer shown above is the chief dimer formed by irradiation of DNA, or from frozen aqueous solutions of thymine or uracil. Other isomers are formed in varying amounts from room temperature solutions of these bases. By clicking on the diagram a drawing showing the pertubation of DNA by this dimerization will be displayed.
In reactions with unsymmetrically substituted alkenes, cyclic enones yield fused cyclobutane rings
with variable regio and stereoselectivity. As shown in the following diagram, the addition of
2-cyclopentenone to propene gives a mixture of regioisomers, both having a cis-fused bicyclo
[3,2,0] heptane unit. In contrast, addition of 2-cyclohexenone to 1,1-dimethoxyethene proceeds
with good regioselectivity and modest stereoselectivity favoring a trans-ring fusion. The last
example demonstrates that unsaturated carbonyl functions other than ketones may lead to
cycloaddition products, and that functional substituents on the alkene moiety may influence the
regioselectivity.
By clicking on the diagram, three more examples will be displayed, The
first two confirm the modest [2+2] regioselectivity noted in the first display and show additional
monocyclic products. The third reaction discloses there is very low retention of configuration in
the cycloaddition of alkene stereoisomers. This stands in marked contrast to the high selectivity
that characterizes [4+2] cycloadditions known as the
Diels-Alder reaction. A similar lack
of stereoselectivity is found in intramolecular [2+2] cycloadditions, as may be seen by clicking
the diagram a second time. Three additional examples of the
intramolecular variant (I. II & III) are also presented in this new display. The importance of
this method for the assembly of complex molecules is shown by the short efficient synthesis of the
sesquiterpene isocomene accomplished by M. Pirrung.
![Enone [2+2] photocycloadditions: cyclopentenone+propene, cyclohexenone+dimethoxyethene, and dihydropyridinone+alkene regiochemistry](/reusch/virtualtext/img/22cyad1.gif)
The poor selectivities and modest quantum yields observed for these cycloaddition reactions have engendered several mechanistic interpretations. Identification of the enone excited state as a π → π* triplet comes from selective quenching studies as well as transient absorption spectroscopy. Bauslaugh's proposal that this triplet adds to the alkene to form a 1,4-biradical proposal is still the simplest and most satisfying explanation of the facts. As shown in the following diagram, the initial bonding may take place at either the α or β-carbon of the enone. The resulting biradicals would also be triplets, and on intersystem crossing to a singlet could yield ground state products by one of three paths.
The latter event accounts for low quantum yields, and is presumed to be fast because isomerization of recovered alkene is small when cis and trans-2-butene are used as the alkene reactant. For the addition of cyclopentenone to isobutene, four possible biradicals may be drawn, the estimated stability order being II > I > III > IV. Finally, although the biradical species are very short lived (ca. 60 ns), it has been possible to trap them by use of the foul-smelling, highly-toxic, hydrogen donor hydrogen selenide. The results of such a trapping study will be displayed below by clicking on the diagram.
Several important conclusions were reached in this case. First, no initial addition to the more substituted carbon of the alkene took place, since tert-butyl substituted cyclopentanones would have been produced (via III & IV) and none were found. Second, although initial bonding at the β-carbon (yielding II) was nearly twice as fast as α-bonding, over 96% of II fragmented to starting materials, compared with 79% of I. Consequently most of the trapped products came from I. These products were formed either by hydrogen atom delivery to both radical sites (giving saturated compounds) or hydrogen delivery followed by hydrogen abstraction by HSe•. (giving unsaturated compounds), as noted in the gray-shaded box bottom-right..
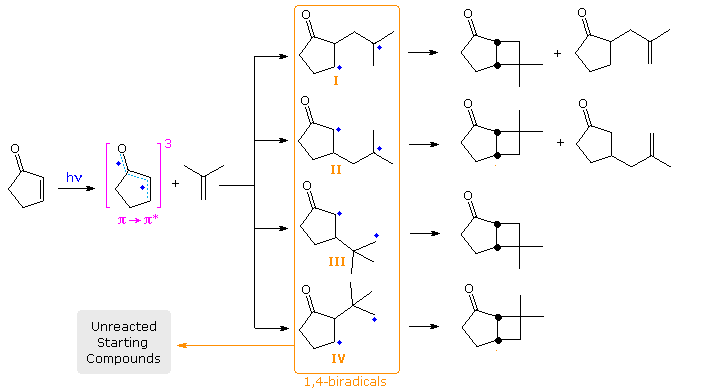
The formation of trans-fused bicyclo [4,2,0] octane products from 2-cyclohexenone has been explained by considering the conformations of the 1,4-biradical intermediates. This will be illustrated above by clicking the diagram a second time. Here, three rotamers (1, 2 & 3) for both the α and β chains are drawn. When the radical sites are far apart (α2 and β3), fragmentation of the biradical is likely. The trans-ring fusion will be formed from conformation α1, and cis from α3. The latter bonding will be subject to some axial hindrance. For the β-manifold, β1 should yield a cis-fused product and β2 the trans-isomer. However the latter bonding would again be hindered.
The de Mayo ReactionIn 1962 Paul de Mayo (Univ. Western Ontario) found that the enol of pentane-1,3-dione gave a 2-acetyl-1-methylcyclobutanol adduct when irradiated in the presence of a cycloalkene. As shown by the top reaction in the following diagram, this cyclobutanol (R1 = R2 = CH3) may then undergo a facile base-catalyzed retroaldol fragmentation to a 1,5-diketone. This sequence thus achieves the attachment of a short alkyl chain to each carbon atom of the double bond. Further base induced reactions, such as epimerization of the initial cis-configuration, and aldol cyclization of the diketone may follow. Following its initial report, this powerful [2+2]-cycloaddition reaction sequence has been put to many uses. One of these is the synthesis of γ-tropolone shown by the bottom equation.
![De Mayo reaction: enone [2+2] cycloaddition then retro-aldol to 1,5-diketones, including gamma-tropolone synthesis](/reusch/virtualtext/img/demayo1.gif)
Two additional applications of the de Mayo reaction in synthesis will be displayed above by clicking on the diagram. The upper reaction is noteworthy for the unexpectedly high regioselectivity of the [2+2] cycloaddition. Ring opening of the bicyclo[3.2.0]heptane moiety proceeds by an ethylagous elimination or Grob fragmentation. Conversion of the resulting bicyclic ketoacetal to β-himachalene was straightforward. The lower reaction demonstrates that poor stereoselectivity in the cycloaddition does not necessarily present a problem. Here, the four new stereocenters in the cycloadduct are lost and/or epimerized by the retroaldol opening of the four-membered ring and subsequent aldol cyclization.
Cyclohexadienone Reactions
Cross-Conjugated DerivativesDerivatives of 2,5-cyclohexadienone are common in nature, and their photochemical transformations posed a challenge to early researchers. Some reactions of 4,4-diphenyl-2,5-cyclohexadienone are presented in the following diagram. Although 2,3-diphenylphenol was a product from irradiation in aqueous dioxane, it is actually formed from 6,6-diphenylbicyclo[3.1.0]hex-3-ene-2-one, the initial rearrangement product. It should be noted that a similar bicyclo[3.1.0]hexane isomer was formed by irradiation of 4,4-diphenyl-2-cyclohexenones, as shown in the gray-shaded box at the bottom right.
![Photochemical rearrangements of 4,4-diphenyl-2,5-cyclohexadienone to bicyclo[3.1.0]hexenone, diphenylphenols and ring-opened acids](/reusch/virtualtext/img/rearrn1.gif)
These photochemical rearrangements occur by way of triplet excited states, which are conveniently depicted as diradicals. Mechanisms for these reactions will be displayed above by clicking on the diagram. Bond reorganization may take place in the triplets, which eventually intersystem cross to singlet species. Charge separation in these states may then lead to rearrangement to a stable product.
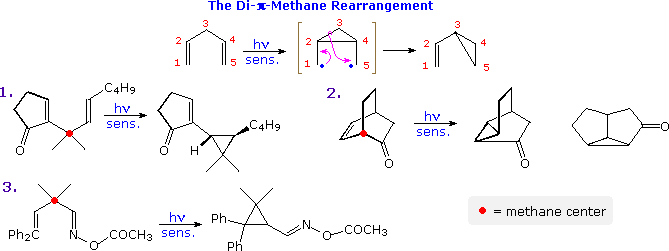
The photo-isomerization shown above is one example of a general family of reactions known as di-π-methane rearrangements, other examples of which are illustrated in the following diagram. These transformations are often photo-sensitized, indicating they proceed by way of triplet excited states. As the name suggests, substrates exhibiting this rearrangement are comprised of two π-functions separated by a saturated (sp3-hybrid) carbon atom (designated by a red dot in the illustration). In the event, a 1,2-shift of one ene-group, followed by a bond formation between the terminal sites of the remaining π-function leads to a vinylcyclopropane product. One of the unsaturated functions may be a carbonyl or imine group, as in examples 2. and 3.
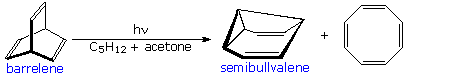
A photo-isomerization of this kind was used by H. Zimmerman (Wisconsin) to prepare the novel self-replicating diene semibullvalene from barrelene.
Conjugated Derivatives
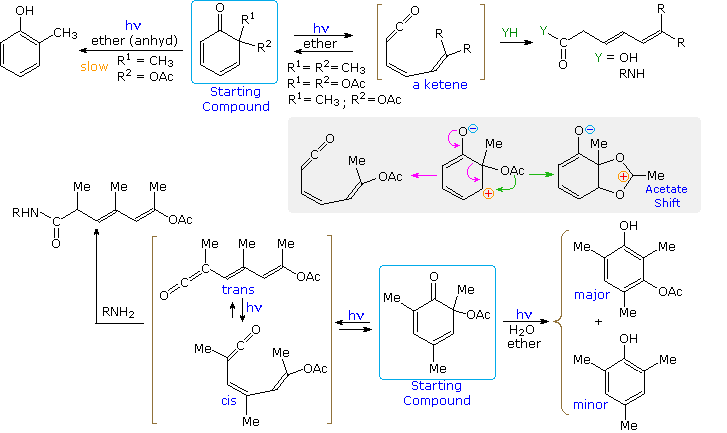
Derivatives of 6,6-disubstituted 2,4-cyclohexadienones are also photochemically reactive. Some examples are given in the following diagram. Electrocyclic ring opening to an unsaturated ketene is the favored transformation. Since nucleophilic compounds such as water, alcohols and amines add rapidly to ketenes, the resulting carboxylic acid or derivative is the final isolated product. If no nucleophilic reactants are present, the conjugated ketene diene will recyclize to the starting compound. Slower reactions leading to phenolic products may then occur. In the last (bottom) example, additional methyl substituents reduce the reactivity of the trans-ketene intermediate, so that only strongly nucleophilic amines are able to trap it. The ketene is removed by light induced isomerization and cyclization. As in the previous dienones, a triplet excited state undergoes decay to polar singlets that are thought to decompose in the manner depicted in the gray-shaded area.
Among the natural dienones displaying interesting photochemical behavior, the sesquiterpene santonin is exceptional, and has been the subject of exhaustive study. Click the button to learn more about this subject.
The names and association (at the time of their major work) of many chemists whose research has led to the development of modern photochemistry are presented in the following table.
| Name | Location |
|---|---|
| D. H. R. Barton | Imperial College, London |
| G. H. Büchi | Massachusetts Institute of Technology |
| O. Chapman | Iowa State University |
| P. de Mayo | University of Western Ontario |
| G. S. Hammond | California Institute of Technology |
| R. S. Liu | University of Hawaii |
| D. C. Neckers | Bowling Green State University |
| J. Saltiel | Florida State University |
| J. C. Scaiano | Ottawa Chemistry Institute |
| G. B. Schuster | Georgia Institute of Technology |
| N. J. Turro | Columbia University |
| P. J. Wagner | Michigan State University |
| A. Weedon | University of Western Ontario |
| N. C. Yang | University of Chicago |
| H. E. Zimmerman | University of Wisconsin |